Прионные болезни — это особый вид тяжелых нейродегенеративных недугов человека и животных. Они характеризуются прогрессирующим поражением головного мозга, в большинстве случаев заканчиваются скорым летальным исходом.
Это особые белковые структуры. Они могут быть как нормальными и входить в состав тканей здоровых людей и животных, так и патологическими, вызывая различного рода заболевания. Еще несколько десятилетий назад считалось, что живая структура обязательно должна содержать так называемые нуклеиновые кислоты — ДНК и РНК. Благодаря им появляется возможность размножаться. Вирусы, грибы, птицы, животные «содержат» нуклеиновые кислоты. Ранее предполагалось, что их отсутствие в тканях означает невозможность полноценного размножения. Прионные белки полностью перевернули эти представления.
Эти молекулы состоят только из белка, но при этом отличаются возможностью размножаться. Проникая в организм, они провоцируют превращение содержащихся в нем нормальных прионов в патологические, тем самым увеличивая свое количество. Этот процесс требует больше времени, чем размножение бактерий или вирусов, поэтому от момента попадания молекулы в организм до развития заболевания может пройти несколько лет.
Прионы и прионные болезни характеризуются высокой устойчивостью. Большинство способов обеззараживания оказываются неэффективны в борьбе с ними. Прионы не погибают при кипячении, могут выдерживать холод до -40 градусов по Цельсию. Они не проявляют чувствительность к УФ-излучению и радиации, сохраняют свои свойства при обработке формалином.
Особенное строение белковых молекул приводит к тому, что организм человека не может с ними бороться. Он не способен продуцировать антитела против прионов, не атакует их лимфоцитами, словно не замечает. Это значит, что проникновение таких молекул в организм человека влечет за собой возникновение того или иного заболевания.
В 1982 году Стэнли Прузинер впервые описал прионные заболевания, за что позднее был удостоен Нобелевской премии.
Задолго до их открытия ученые в своих работах исследовали ряд патологий человека и животных, причину которых долгое время не могли установить. В XVIII веке на территории Великобритании была зарегистрирована «почесуха» овец. Животные страдали от сильного зуда, нарушения движения и припадков, которые указывали на поражение ЦНС. В 1957 году Карлтон Гайдушек описал недуг у племени Фор, жители которого обитали в высокогорьях Папуа— Новой Гвинеи. Патология была связана с каннибализмом и передавалась от одного человека к другому.
С 1986 года в Англии, а затем и во многих других странах, ученые зафиксировали несколько вспышек заболевания, получившего позднее название «коровье бешенство». Оно преимущественно поражало крупный рогатый скот. «Коровье бешенство» через небольшой отрезок времени приобрело масштабы эпидемии, а причиной его возникновения стали прионы. В 90-е годы специалисты доказали передачу этого недуга человеку вместе с молоком и мясом крупного рогатого скота.
В настоящее время подробное изучение заболеваний с невыявленными причинами поспособствовало тому, что ученые высказали ряд предложений относительно прионной природы развития. К их числу относится патология Крейтцфельдта-Якоба, болезнь Альцгеймера. Симптомы и признаки этих недугов имеют много общего. Несмотря на масштабные успехи в изучении этих расстройств, многое еще остается за гранью постижимого.
В современной медицине выделяют три способа заражения.
- Трансмиссивный. Прионы передаются от одного вида млекопитающего к другому. Ранее говорилось о существовании так называемых межвидовых барьеров. Это значит, что передача от коровы к человеку невозможна. Сегодня ученые опровергают эту точку зрения. Белковые молекулы могут передаваться от инфицированного животного или человека. Причины возникновения прионных болезней обусловлены употреблением в пищу мяса/молока зараженного животного, использование его биологических тканей (пересадка роговицы, препараты крови и т. д.). Различные биоматериалы обладают неодинаковой степенью патогенности. Наибольшую заразность проявляют ткани мозга, следующую ступень занимают кровь и ее препараты.
- Наследственный. Заболевание развивается на фоне генной мутации, формирующейся в области 20-й хромосомы. Именно этот участок отвечает за наличие нормального прионного белка. Его функционирование до сих остается малоизученным. В случае генных мутаций вместо здорового приона синтезируется патологический, что неминуемо приводит к развитию недугов.
- Спорадический (спонтанное появление аномального белка).
Таким образом, прионные болезни могут носить как наследственный, так и инфекционный характер. Вне зависимости от способа проникновения аномального белка в организм, он может стать причиной заражения других людей.
Патологические белки отличаются способностью вызывать губчатые энцефалопатии, то есть поражение ЦНС. С морфологической точки зрения это означает формирование в клетках мозга полостей, гибель нейронов, разрастание на их месте соединительной ткани и конечную атрофию мозга. На фоне скопления прионов наблюдается формирование амилоидных бляшек. Все эти процессы возникают при отсутствии явных признаков воспаления.
На сегодняшний день ученые могут точно назвать несколько недугов, причиной развития которых являются аномальные белковые структуры:
- болезнь Крейтцфельдта-Якоба;
- болезнь куру;
- болезнь Альперса (прогрессирующая спонгиозная энцефалопатия);
- семейная фатальная инсомния;
- болезнь Герстманна-Штреусслера-Шейнкера.
Далее рассмотрим каждую патологию более подробно.
Болезнь Крейтцфельдта-Якоба отличается своим разнообразием, поэтому специалисты разделели ее на несколько форм:
- спорадическая;
- семейная;
- ятрогенная;
- новая атипичная форма.
Спорадический вариант заболевания ранее считался самым распространенным. Первые его симптомы появляются в возрасте после 55 лет. Однако за последние несколько лет статистика изменилась. После появления сведений об эпидемии «коровьего бешенства» все чаще стали фиксировать случаи атипичной формы вследствие заражения КРС. Для этого вида характерно ранее появление. В большинстве случаев страдают молодые люди. Симптомы заболевания подразделяются на две условные группы: неврологические и психические. Первоначально у заразившихся отмечается головная боль, нарушение сна, снижение аппетита. Постепенно к этим симптомам прибавляется ухудшение памяти, потеря зрения. Психические нарушения проявляются в форме галлюцинаций и бреда. Заболевание отличается стремительным развитием, в последней стадии характеризуется полной обездвиженностью тела. Человек утрачивает контроль над функцией тазовых органов. С таким диагнозом люди живут не более двух лет.
Появление семейной формы обусловлено мутациями на генном уровне в зоне 20-й хромосомы. Заболевание отличается аутосомно-доминантным характером. Первые признаки возникают примерно на 5 лет раньше, чем при спорадической варианте.
Ятрогенная форма развивается вследствие инфицирования человека во время хирургического вмешательства. Статистическая информация по этому варианту недуга отсутствует, поскольку сложно доказать патогенез прионных болезней. Продолжительность инкубационного периода варьируется от 7 месяцев до приблизительно 12 лет. Он определяется совокупностью нескольких факторов: способа проникновения аномальных белков в организм, их количества, исходного генотипа человека. Быстрее всего недуг развивается при непосредственном проникновении прионов в ткани мозга в результате оперативного вмешательства. Больше времени требуется при инфицировании на фоне трансплантации роговицы или твердой мозговой оболочки. У пациентов постепенно развивается мозжечковая атаксия, нарушение речи и мышечного тонуса, деменция.
«Коровье бешенство» стало приобретать актуальность после эпидемии у КРС в 90-х годах. Прионная болезнь, симптомы которой проявляются в возрасте от 30 до приблизительно 40 лет, смертельна для человека. Как и при ятрогенном варианте, неврологические признаки преобладают над психическими.
Это заболевание аутосомно-доминантного характера, которое передается исключительно по наследству. Фатальная инсомния встречается редко. Она известна в науке начиная с 1986 года. Первые ее симптомы проявляются в возрасте от 25 лет до приблизительно 71 года.
Эпидемиология прионных болезней этого типа малоизученна. Главным признаком семейной фатальной инсомнии является бессонница. Организм постепенно утрачивает возможность полноценно регулировать фазы бодрствования и сна. Также у пациентов появляются нарушения двигательной активности и мышечная слабость. Известны случаи вегетативных расстройств, которые проявляются повышением АД, чрезмерным потоотделением. Из психических нарушений можно отметить панические атаки, зрительные галлюцинации и кратковременные эпизоды спутанности сознания. Вследствие постоянной бессонницы наступает истощение организма, больной умирает.
Инфекционные формы прионов были детально изучены благодаря этому заболеванию, точнее, племени людоедов. До 1956 года среди жителей Папуа — Новой Гвинеи были распространены традиции так называемого ритуального каннибализма — употребление в пищу мозга умершего человека. Считается, что у одного из членов этого племени возникла инфекция, которая впоследствии распространилась на других людей после совершения ритуала. С момента отмены этой традиции случаи заболеваемости стали фиксироваться в несколько раз реже, сегодня этот недуг практически не встречается.
Продолжительность инкубационного периода составляет от 5 до приблизительно 30 лет. Именно поэтому болезнь Куру относят к категории «медленные вирусные инфекции». Недуг проявляется мозжечковыми расстройствами вместе с неконтролируемым смехом, дисфункцией глотания и мышечной слабостью. В терминальной стадии развивается деменция. Люди с таким диагнозом живут не более 30 месяцев.
Заболевание преимущественно развивается у детей младшего возраста (до 18 лет). Недуг передается по аутосомно-рецессивному типу, в случае совпадения двух патогенных генов папы и мамы. Среди основных симптомов можно выделить нарушение зрения и эпилептические припадки. В медицинских справочниках встречаются описания острых состояний заболевания, протекающие по типу инсульта. Болезнь Альперса также характеризуется поражением печени, которое быстро перерастает в хронический гепатит и заканчивается циррозом. Пациенты умирают из-за интоксикации организма в течение 12 месяцев с момента диагностирования первых симптомов.
Данный вариант заболевания причисляется к наследственному типу. Встречается очень редко (один случай примерно на 10 000 000 населения). Появление первых признаков обычно отмечают у пациентов после 40 лет. Развитие синдрома начинается с мозжечковых расстройств. Первоначально появляется головокружение. По мере развития недуга прогрессируют координаторные нарушения, постепенно самостоятельное передвижение становится невозможным. Наряду с перечисленными симптомами появляются нарушения тонуса мышц, наблюдается снижение зрения и слуха, проблемы с глотанием и звуковоспроизведением. В терминальной стадии врачи фиксируют проявления деменции. Продолжительность жизни больных с таким диагнозом составляет до 10 лет.
Синдром Альцгеймера и болезнь Паркинсона, симптомы и признаки которых имеют общую природу, развиваются по схожему с прионными недугами пути. Молекулы бета-амилоида, белка тау и прочие структуры так же формируют отложения патогенной природы в мозговых тканях. Однако, заразиться этими недугами невозможно. Это значит, что амилоидные фибриллы формируются за счет испорченных белковых молекул, но на «здоровые» действие «больных» не распространяется.
Совсем недавно ученые провели ряд исследований на мышах, которые опровергли это предположение. После введения патогенных белков в мозг абсолютно здорового животного у него появлялись характерные амилоидные бляшки. Это значит, что болезнетворный белок все же может заражать здоровые структуры. Данное открытие принадлежит специалистам из Техасского университета. В ближайшем будущем выйдет еще одна работа ученых из Лондона, которая доказывает, что болезнь Альцгеймера, симптомы и признаки недуга в полной мере могут передаваться от одного человека к другому.
Напомним, что болезнь Паркинсона характеризуется постепенной гибелью нейронов, вырабатывающих медиатор дофамин. Из-за этого у человека нарушается регуляция движений и мышечного тонуса, что проявляется тремором, общей скованностью. От паркинсонизма страдает каждый сотый человек, перешагнувший шестидесятилетний рубеж. Заболевание начинает свое развитие с замедленности движений, что особенно заметно, когда человек одевается или принимает пищу. Впоследствии нарушается речь, глотательные рефлексы. К сожалению, сегодня медицина не может рекомендовать эффективное лечение людям с диагнозом «болезнь Паркинсона». Симптомы и признаки этого недуга можно смягчить благодаря симптоматической терапии. Однако большая часть таких препаратов вызывает ряд побочных эффектов.
Синдром Альцгеймера — это заболевание, характеризующееся отмиранием нейронов, в результате чего у пациентов развивается старческое слабоумие. Первые симптомы этого недуга могут проявиться уже в возрасте 40 лет. Его диагностируют в большинстве случаев у малообразованных людей. У человека с высоким уровнем интеллекта больше шансов справиться с проявлениями Альцгеймера ввиду многочисленных связей между нейронами.
Заболевание начинает свое развитие с нарушений памяти. Первичная стадия обычно остается незамеченной для окружающих. Начальные симптомы часто пытаются скрыть или списывают на стрессы и чрезмерную загруженность на работе. По мере прогрессирования недуга клиническая картина видоизменяется. Больной перестает ориентироваться в пространстве, из его памяти выпадают приобретенные ранее навыки письма, чтения. Сначала забываются ближайшие по времени события. Когда патология начинает прогрессировать, необходимо использовать любую возможность для поддержания способности человека к самостоятельному обслуживанию, постараться предупредить возникновение депрессии. В решении этой проблемы может помочь более сильный слуховой аппарат или правильно подобранные очки. Специфического лечения синдрома Альцгеймера не существует. При появлении первичных ее симптомов важно пройти полное обследование у невролога. Специалисты для лечения обычно рекомендуют препараты, облегчающие течение недуга и сдерживающие его развитие.
Специфических мер диагностики в настоящее время не представлено. Например, похожие результаты ЭЭГ, что и при болезни Крейтцфельдта-Якоба, бывают при других патологиях мозга. МРТ характеризуется невысокой диагностической значимостью, так как у 80% обследованных выявляют неспецифические сигналы. Однако, данное исследование позволяет распознать атрофию мозга. Ее выраженность усугубляется по мере того, как прогрессируют прионные болезни человека.
Дифференциальная диагностика проводится со всеми патологиями, одним из проявлений которых является деменция (болезнь Альцгеймера, васкулиты, нейросифилис, герпетический энцефалит и другие).
К сожалению, в настоящее время все прионные болезни неизлечимы. Пациентам назначается симптоматическая терапия с использованием противосудорожных средств, которая только облегчает страдания. Прогноз неутешителен. Все известные заболевания прионной природы смертельны для человека.
В настоящее время ученые со всего мира занимаются активным поиском универсального лекарства. Проводятся исследования с использованием животных. Предполагается, что в борьбе с такими недугами впоследствии будут применяться стволовые клетки, а также самые обычные дрожжи. Экспериментальные препараты в настоящее время не отличаются высокой эффективностью, поэтому их назначение считается нецелесообразным.
От развития спорадических и наследственных вариантов прионных заболеваний обезопасить себя практически невозможно. Некоторые патологии можно исключить, пройдя специальную генетическую экспертизу. Однако это в нашей стране весьма затруднительно, поскольку лаборатории, выполняющие подобного рода диагностику, находятся преимущественно за рубежом.
В случае наследственных заболеваний перед беременностью рекомендуется проконсультироваться с врачом-генетиком. Это поможет в будущем избежать проблем со здоровьем ребенка.
Для того чтобы обезопасить себя от болезни Крейтцфельдта-Якоба, рекомендуется отказаться от употребления в пищу мяса из регионов, где зафиксированы случаи заболевания крупного рогатого скота. В первую очередь речь идет о европейских странах. Также не следует использовать в лечении препараты, изготовленные из крови животных или человека. Лучше их заменять синтетическими аналогами.
Прионные болезни — это недостаточно исследованные формы инфекционных и наследственных поражений, которые возникают в организме человека на фоне проникновения аномальных белков. В большинстве случаев они поражают ЦНС. Клиническая картина отличается схожей симптоматикой. Первоначально у человека пропадает аппетит и зрение, нарушается координация в пространстве. На заключительной стадии развивается деменция, когда больной не может самостоятельно о себе позаботиться. Результат любого заболевания всегда одинаковый — смерть. В настоящее время медики не располагают эффективными средствами против патологий такой природы.
источник
Прионная болезнь — та, о которой в России слышали только врачи, и, быть может, редкие родственники пациентов. На самом деле она входит в список самых опасных заболеваний в мире, и по счастью, не передается воздушно-капельным путем, иначе бы спасения не было вообще. Болезнь заразна, смертельна в 100% случаев, чаще всего быстротечна. И в России такой диагноз ставить запрещено, правда, официального запрета не существует. В итоге пациенты оказываются без диагноза, не зная от чего умирают, родственники не понимают, что они в огромной опасности. А обычные люди даже не представляют, чем может обернуться съеденный стейк с кровью, блюдо из коровьих мозгов или пробование сырого фарша после соления. «Хайтек» совместно с Ойбеком Тургунхужаевым, руководителем отделения нейрореабилитации в Междисциплинарном центре реабилитации, разбирался, почему нам стоит бояться прионов и кто находится в неотвратимой группе риска.
С появлением Интернета и свободных СМИ, люди стали узнавать всё больше о смертельных заболеваниях — инфекционных, вирусных, онкологических и наследственных. Но мало кто слышал о фатальных прионных болезнях. Несмотря на клинические испытания, на данный момент не существует ни одного доказанного универсального лечения этой группы заболеваний. Невролог Ричард Джонсон из Университета Джона Хопкинса говорит, что если прионы пациента превратились в патологические, он умирает, и мы не можем этого избежать.
Прионные заболевания — они же называются трансмиссивными губчатыми энцефалопатиями — представляют собой семейство редких прогрессирующих нейродегенеративных заболеваний, которые поражают как людей, так и животных. Их отличают:
- длительный инкубационный период;
- характерные губчатые разрыхления мозговой ткани, связанные с потерей нейронов;
- неспособность иммунной системы отреагировать на заражение, инициируя воспалительный процесс.
Прионные болезни поражают как людей, так и животных, быстро прогрессируют и всегда приводят к летальному исходу.
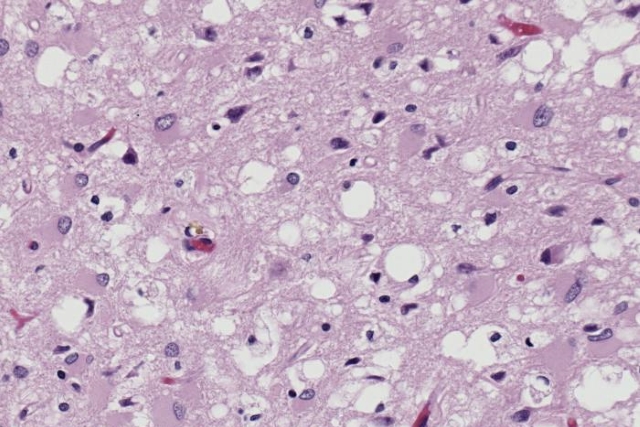
Возбудитель болезни — прионы — тип белков с аномальной третичной структурой, не содержащий нуклеиновых кислот. Сам термин относится к патологическим патогенным агентам, которые способны вызывать аномальное сворачивание специфических нормальных клеточных белков, которые называются как раз прионными белками, встречающихся чаще всего в мозге. Функции этих нормальных прионных белков до сих пор полностью не изучены.
Болезнь Крейтцфельдта – Якоба — БКЯ, псевдосклероз спастический, синдром кортико-стриоспинальной дегенерации, трансмиссивная спонгиоформная энцефалопатия, коровье бешенство.
Это прогрессирующее дистрофическое заболевание коры большого мозга, базальных ганглиев и спинного мозга. Считается основным проявлением губчатой энцефалопатии (прионной болезни). Излечение невозможно. БКЯ поражает людей всех национальностей и рас, мужчин и женщин, взрослых и детей.
Прионные белки — нормальные белки, которые присутствуют у каждого человека. Но есть определённые группы людей, у которых присутствует генетическая мутация, предрасполагающая их к синтезу патогенного прионного белка. Прионные болезни также могут передаваться путём прямого заражения, передача может произойти и в ходе хирургических манипуляций, использования человеческого гормона роста или употреблении заражённого мяса. Такой вид заражения называется ятрогенным и он остаётся в процентном меньшинстве относительно иных форм БКЯ.
Процентное соотношение ятрогенных случаев болезни Крейтцфельдта – Якоба в исследовании National CJD Research & Surveillance Unit у 177 пациентов.
- Гормон роста (соматотропин) — 53,1% (94 случая).
- Твердая мозговая оболочка (в том числе поедание) — 38,9% (69 случаев).
- Гонадотропный гормон — 2,25% (4 случая).
- Нейрохирургический инструментарий — 2,25% (4 случая).
- Пересадка роговицы — 1,69% (3 случая).
- Электроды для стереоэлектроэнцефалографии — 1,12% (2 случая).
- Пересадка печени — 0,56% (1 случай).
Есть случаи заражения, которые не классифицируются ни по одной из двух вышеупомянутых причин, в таком случае они считаются спорадическими, то есть возникшими спонтанно и самостийно, по независящим от генетики или внешних факторов обстоятельствам.
Доктор Ойбек Тургунхужаев, руководитель направления нейрореабилитации Междисциплинарного центра реабилитации (Москва), говорит, что окончательный диагноз человеку с подозрением на какое-либо прионное заболевание основывается на оценке клинических признаков и симптомов и ряде вспомогательных исследований. Долгое время единственным методом подтверждения диагноза была электроэнцефалография. Но поскольку общая чувствительность этого метода ограничена, полезность этого исследования была поставлена под сомнение.
Прионные заболевания неизлечимы, они неизбежно фатальны. Кроме этого, проблема заключается в том, что для постановки достоверного диагноза необходимо проводить вскрытие. Любое вскрытие — это риск для патологоанатома, так как были случаи ятрогенного заражения специалистов от умерших пациентов.
По приказу Роспотребнадзора, о том, что человек заболел прионной болезнью необходимо извещать в течение двух часов. При этом установление такого диагноза ведет за собой, по российским инструкциям, утилизацию всего оборудования, с которым пациент был в контакте. Именно поэтому, когда Медуза рассказывала случай одной из больных БКЯ, все клиники говорили о том, что у них нет оборудования для наблюдениях таких пациентов. На самом деле — это просто способ не потерять миллионы рублей, утилизируя даже аппарат МРТ. Если бы речь шла о сотнях поставленных диагнозов прионной болезни (например, в США регистрируется 300 случаев ежегодно, возможно, их больше), тогда речь шла бы о потери миллиардов рублей для российских больниц и бюджета. Именно поэтому официально диагноз не ставится, врачи не хотят об этом говорить, так как никакого официального распоряжения не существует, что диагноз ставить нельзя. В итоге выходит, что заболевание есть, смерти есть, а причины для родственников и умирающих людей — нет.
Им никто не скажет, что скорее всего родственники уже заразились. Никто не скажет, что нельзя пробовать сырой фарш или есть сырое мясо, тем более мозги. Также как из-за того, что диагноз не ставится, можно случайно пересадить орган больного прионной болезнью, тем самым заразив другого человека. Также это может произойти через хирургический инструмент (такие случае были, об этом ниже).
Когда мы просили хоть кого-то рассказать нам о прионных болезнях, практически никто не готов был говорить открыто. Так мы анонимно поговорили с врачом-неврологом одной из крупнейших московских больниц. «С прионами две проблемы. Во-первых, для постановки достоверного диагноза необходимо проводить вскрытие. Хотя формально (по российским руководствам, например) проводить вскрытие можно, хотя и в особенных условиях. Любое вскрытие — это, естественно, дополнительный риск для патологоанатомов, потому что были описаны случаи заражения патологоанатомов от умерших пациентов. никто не хочет переводить на них риск.
Во-вторых, так как прионные инфекции — это тяжело протекающие, неизлечимые заболевания (хотя и с довольно сложным путем передачи), в нашей стране чертовски сложные законы для регистрации и ведения таких пациентов; о случаях обязаны сообщать в случае выявления чуть ли не в течение двух часов, после постановки диагноза необходимо проводить уничтожение части в том числе дорогостоящего оборудования, которое, как может оказаться по факту, даже рядом не лежало с пациентом, нужно переоформлять документы и так далее.
Довольно грустный, но тем не менее правдивый, факт заключается в том, что правильный диагноз не имеет значения для пациента. Лечения до сих пор нет».
Болезнь Крейтцфельда – Якоба (БКЯ) является одной из разновидностей прионных болезней. Это быстро прогрессирующее, фатальное нейродегенеративное заболевание, которое, как полагают, вызвано аномальной изоформой прионного белка. БКЯ встречается во всем мире, и согласно статистике, во всем мире заболевает 1 из миллиона человек.
Прионные болезни не идут по одному и тому же сценарию, у людей, страдающих одним и тем же прионным поражением могут разниться эпидемиология и патогенез. Болезнь Крейтцфельда-Якоба делят на несколько типов.
Спорадическая Болезнь Крейтцфельда-Якоба (сБКЯ) — наиболее распространенный вид трансмиссивных губчатых энцефалопатий человека, на долю которого приходится около 85% случаев зарегистрированных заболеваний прионной природы. СБКЯ имеет очень быстрое течение болезни — средняя продолжительность жизни после проявления признаков составляет всего шесть месяцев. Более 90% пациентов умирают в течение года после появления симптомов. Пик заболеваемости приходится на пожилых людей возрастом 60–70 лет, в других возрастных группах случается куда реже. Одной из гипотез происхождения сБКЯ является мнение, что это спонтанное нейродегенеративное заболевание, возникающее в результате соматической мутации гена PRNP или случайного структурного изменения в белке PrP, вызывающее образование PrPSc2. Эпидемиологические исследования не выявили связи спорадической формы БКЯ с экологическими факторами.
Первые симптомы сБКЯ обычно неспецифические: головная боль, недомогание, кашель, головокружение и изменение поведения, настроения или провалы в памяти. Для подтверждения диагноза должно пройти время, чтобы появились и иные основания полагать прионную природу. Классическими клиническими признаками с БКЯ являются:
- быстрое снижение когнитивных способностей;
- атаксия (нарушение согласованности движения различных мышц);
- миоклонус (быстрые внезапные сокращения отдельных мышц), оканчивающиеся акинетическим мутизмом (торможение всех двигательных функций, кроме фиксирующих движений глазных яблок).
Окончательный диагноз зависит от оценки клинических проявлений и результатов лабораторных тестов.
Акинетический мутизм — состояние при котором пациенты перестают двигаться и следить глазами за целью, за исключением реакции глаз на раздражители или длительной фиксации взгляда, их мышцы самостоятельно или под воздействием внешних факторов периодически быстро сокращаются. Пациенты страдают недержанием, не издают никаких звуков или только нечленораздельные шумы. Если глотание сохраняется, пациенты могут прожить в этом состоянии в течение нескольких недель, даже годы при иных благоприятных факторов, получая питание внутривенно или через трубку. При спорадической форме БКЯ, пациенты доходят до этого состояния в течение первых недель заболевания. Всамых стремительных сценариях, 10% пациентов доходят до этого состояния за год.
Долгое время самым информативным способом постановки диагноза было проведение диффузионно-тензорной МРТ. Этот способ является наиболее доступным, относительно неинвазивным и действенен при ранних изменениях в коре головного мозга. СБКЯ можно обнаружить через маркеры в назальных слизистых оболочках, спинномозговой жидкости, моче или крови, но эти тесты часто дают ложноположительные результаты — белок 14.3.3 не является специфичным без появления сопутствующей клинической картины. Протеинограмма является новым оптимальным методом диагностирования сБКЯ, так как она самая чувствительная из всех вышеназванных.
Белки 14-3-3 — семейство регуляторных молекул, встречающихся у всех эукариот. Они связываются со множеством других белков, регулируя их функции и тем самым влияя на множество процессов, в том числе регулировку клеточного цикла, контроль метаболизма, апоптоз, контроль транскрипции генов. Они были обнаружены более 40 лет назад при систематической классификации белков нервной ткани, где их содержание превышает 1% от всех белков. К настоящему времени описано более 300 различных белков-мишеней, способных взаимодействовать с 14-3-3.
Протеинограмма — исследование, изучающее количественное соотношение разновидностей белка в крови. В понятие общего белка входят все возможные белки, несмотря на их различия в строении и функциях.
Наследственная форма болезни Крейтцфельда – Якоба и также связанная с генетическими мутациями фатальная семейная бессонница составляют всего 10% от всех случаев прионных болезней. Множество исследований указывает на то, что общий путь в патогенезе заболевания может быть общим как для спорадических, так и для наследственных форм прионного заболевания, за исключением того, что в первом случае превращение белка происходит без участия каких-либо факторов, а не предопределено наличием мутации в генах.
Исследования наследственной формы БКЯ и фатальной семейной бессонницы предоставили учёным возможность изучить течение фенотипической гетерогенности (разнообразие «штаммов») прионной болезни. Хотя многие другие нейродегенеративные заболевания, такие как болезнь Альцгеймера, боковой амиотрофический склероз и хорея Хантингтона, достаточно похожи по фенотипу, прионная болезнь включает в себя множество клинически различных признаков.
Фенотип — совокупность характеристик, присущих на определённой стадии развития болезни. Фенотип формируется на основе генотипа.
Полиморфизм — способность некоторых организмов существовать в состояниях с различной внутренней структурой или в разных внешних формах.
При наследственном прионном заболевании, фенотип заболевания будет определяться комбинированным эффектом патогенных мутаций, полиморфизма кодонов 129 и типа PrPSc. Полиморфизм кодона 129 играет двойную роль в прогнозировании исхода заболевания. Главным в понимании патогенеза прионной болезни является детальное и точное знание процессов и условий in vivo для образования PrPSc, которые неизбежно приводят к развитию и выражению заболевания. Эти знания позволят разработать рациональную и эффективную стратегию терапевтического вмешательства.
Новый вариант болезни Крейтцфельда – Якоба (nvCJD) был впервые идентифицирован в 1996 году. Последующие исследования подтвердили гипотезу, что эта форма связана с бычьей губчатой энцефалопатией. Скорее всего, пациенты употребляли в пищу мясо, содержащее патологические прионы мозга коров.
В отличии от спорадической формы, болезнь не имеет четкого возраста заражения. У пациентов с нБКЯ часто выявляют и психиатрические симптомы, потому порой она ошибочно диагностируется как психическое, а не неврологическое расстройство. Истинная причина психиатрических симптомов кроется в когнитивных нарушениях, постоянных болях в конечностях, нарушениях адекватности ощущений (парестезия или дизестезия), расстройствах речи или зрения.
В течение 6–8 месяцев развиваются пороки управления мышечной системой, но в некоторых случаях развитие болезни может длиться и более 18 месяцев. Потому этот диагноз достаточно трудно поставить при появлении первых признаков заболевания. Если у пациента возникают неконтролируемые движения, возрастает вероятность грамотного диагностирования нБКЯ. В отличие от спорадической формы, где характерны внезапные мышечные спазмы при напряжении (миоклонус), в случае с новой формой возможны и дистония (синдром, при котором происходит постоянное спазматическое сокращение мышц), и хорея (синдром, характеризующийся беспорядочными, отрывистыми, нерегулярными движениями).
Летальная стадия новой формы похожа на летальную стадию спорадической формы болезни Крейтцфельда-Якоба, она проходит с прогрессирующей потерей контроля над мышцами, часто приводящей к состоянию акинетического мутизма.
В отличие от более распространенных слабоумных состояний, которые обычно развиваются годами, быстро прогрессирующие деменции могут развиваться в течение нескольких месяцев, недель или даже дней и приводить к смерти. На спорадическую форму БКЯ приходится 46,9 % всех зарегистрированных случаев быстро прогрессирующей деменции, на генетическую форму прионных заболеваний — 13,6%. 39% всех случаев составляют лобно-височная деменция (FTD), кортикобазальная дегенерация (CBD), болезнь Альцгеймера (AD), деменция с тельцами Леви (DLB) и прогрессивный паралич.
Как правило, спорадическая форма БКЯ представлена совокупностью деменции и нейродегенеративных или психиатрические симптомов. У таких больных распространены пирамидная, мозжечковая и фокальная кортикальная дисфункция. У трети пациентов деменции предшествуют жалобы на усталость, головную боль, нарушение сна, недомогание, потерю веса, боль, депрессию или изменения в поведении.
Неврологические симптомы, включая атаксию, дизестезию, слабоумие или мышечные расстройства (хорея, миоклонус или дистония) появляются позже. Большинство случаев быстро прогрессирующей деменции без других сопутствующих симптомов случается у пожилых людей из-за метаболических нарушений или острых инфекций (пневмонии или инфекции мочевыводящих путей). Потому перед прохождением лабораторных тестов врачи первостепенно указывают быстро прогрессирующую деменцию без явного диагноза. Окончательный вердикт будет зависеть от полученных результатов анализов и обследований, заболевания будут отличаться в зависимости от клинической картины. ЭЭГ может помочь исключить судорожную активность головного мозга и обратиться к диагностике других состояний, таких как БКЯ.
Быстро прогрессирующая деменция представляют собой одну из самых сложных неврологических проблем. Дифференциальная диагностика широко используется для подтверждения окончательных диагнозов, которые могут относиться к нейродегенеративным, аутоиммунным, инфекционным и опухолевым заболеваниям. Даже при таком тщательном подходе к обследованию пациентов, небольшой процент случаев диагностируются уже после смерти.
Фатальная семейная бессонница — это редкое прионное заболевание, которое в буквальном смысле лишает сна и приводит к снижению всех нейро-двигательных и психических функций. Можно выделить две формы этой болезни: генетическую и спорадическую.
Генетическая форма связана с мутацией, приводящей к превращению белка PrP в прионный белок. Спорадическая же появляется спонтанно, без каких-либо предпосылок. Это заболевание отличается от других прионных заболеваний областью поражения — превращение прионных белков в патологические преимущественно происходит в одном отделе головного мозга — таламусе, который, в том числе, отвечает и за сон.
Средний возраст появления симптомов при генетической форме фатальной семейной бессонницы — 40 лет. Ранние симптомы выражаются в виде незначительных трудностей с засыпанием и сном, мышечными подергиваниями, судорогами и оцепенение. Во время сна люди могут неспокойно спать: двигать ногами и руками, ворочаться. В конце концов, больные перестают спать. В силу этого снижается умственная активность, утрачивается мышечная координация (атаксия). У больных прослеживается повышение артериального давления, учащение пульса и избыточное потоотделение. В отличие от других прионных болезней при фатальной семейной бессоннице нет губкообразной дегенерации вещества головного мозга. Основными признаками поражения являются дегенерация нейронов и реактивный глиоз (образование «рубцов» на и в мозговой ткани) в ядрах таламуса, а также дегенерация нейронов в комплексе ядер продолговатого мозга.
Диагноз фатальной семейной бессонницы в генетической форме подтверждается генетическим тестированием. В случае со спорадическими случаями, могут обнаружить нарушения в структуре сна и аномалии в таламусе полисомнография и позитронно-эмиссионная томография (ПЭТ). Средняя продолжительность жизни с начала первых симптомов заболевания — 3 года, лечения не существует.
Синдром Герстманна-Штраусслера-Шейнкера — это редкая генетическая форма трансмиссивной губчатой энцефалопатии, которая впервые была описана австрийскими неврологами в 1936 году. Синдром чаще всего проявляется в возрасте 40–50 лет, и вызван мутациями генов прионного белка PRNP на 20 хромосоме.
Клиническая картина синдром схожа со спорадической формой БКЯ, но он отличается продолжительностью и медленно прогрессирующей деменцией наряду с иными симптомами. Синдром Герстманна-Штраусслера-Шейнкера может длиться как нескольких месяцев так и несколько лет, средняя продолжительность жизни — 5 лет. Диагностировать заболевание можно даже на ранних стадиях посредством проведения магнитно-резонансной томографии. На МРТ будут наблюдаться губчатые изменения в коре и разрастание глиальных клеток.
Глиальные клетки или нейроглия — совокупность вспомогательных клеток нервной ткани. Составляет около 40 % объёма ЦНС. Количество глиальных клеток в мозге примерно равно количеству нейронов.
Глиальные клетки имеют общие функции и, частично, происхождение (исключение — микроглия). Они составляют специфическое микроокружение для нейронов, обеспечивая условия для генерации и передачи нервных импульсов, а также осуществляя часть метаболических процессов самого нейрона.
Обнаруживать прионную болезнь достаточно трудозатратно, учитывая то, что если диагноз подтвержден, пациенту уже ничем не помочь, а больницы теряют миллионы рублей. И самое жуткое, что все люди на Земле в группе риска. Прионы не пощадят никого. Поэтому не пробуйте фарш, после того, как его посолили. Не ешьте потроха и мозги коров и свиней, и посещайте врача вовремя. В конце концов, МРТ врать не будет.
источник
Прионные заболевания одни из самых опасных для человека. Болезнь Крейтцфельдта Якоба (БКЯ) — это нарушение функционирования мозга, которое прогрессирует стремительно и приводит к снижению интеллектуальных способностей пациента. Существенный вклад в изучение патологии внесли немецкие ученые, в честь которых она и получила свое название.

Эта прионная энцефалопатическая болезнь лидирует среди подобных недугов. На нее приходится 85% всех пострадавших людей, без разделения по расам, половому признаку или возрасту. Чуть больше болезней куру у женщин Новой Гвинеи, т. к. у них практикуется ритуальный каннибализм, при котором потребляется мозг умерших. При этом в их организм попадает огромная доза прионов.
Первые признаки недуга проявляются в среднем возрасте ближе к пятому десятку, но зарегистрированы случаи заражения и в более раннем периоде. Классическая форма болезни имеет усредненный возраст в 63-64 года, а новая — 28-30 лет.
Недуг Крейтцфельдта Якоба носит инфекционный характер. Заражение возможно несколькими путями, при:
- трансплантации инфицированных органов и тканей;
- использовании нейрохирургического инструмента без должной обработки;
- введении препаратов крови, натуральных гормонов, например, для лечения бесплодия Кортексином и т. п.
Новая форма недуга поражает человека при употреблении в пищу зараженного прионами мяса коровы, овец, коз. Исследования показали, что если инфекционный белок попал в организм, то болезни БКЯ не избежать. При этом в головном мозге человека есть прион, но с другим строением.
Инфекционный белок при проникновении в организм не разрушается, а по кровотоку попадает в мозг, где засоряет поверхность нейронов. Он вступает в реакцию с нормальными прионами, превращая их в патогены. Это приводит к бляшкам и гибели нейронной системы.
Такие болезни имеют длительный инкубационный период, т. к. инфекционным прионам нужно время для проникновения в мозг и трансформацию нормальных тканей в патогенные. Как долго это будет, зависит от того, как заразился человек. Например, при их занесении в мозг через хирургический инструментарий болезни хватит полутора лет.
Если это трансплантация, то 5-6 лет, через лекарственные препараты — 12. Возможна передача болезни генетически, при которой патологические прионы образуются сами по себе.
Классификация этой болезни состоит из 4 основных видов, каждый из которых имеет свой путь заражения, симптоматику и инкубационный период.
Ориентируясь на прионную теорию, спонтанная форма БКЯ возникает в мозгу человека, не имея на то никакой внешней причины. В группе риска 50-летние люди. Но вероятность пострадать от нее всего пару случаев на миллион. Первые признаки — это краткие провалы в памяти, перепады настроения, потеря интереса ко всему.
Больному неважны любые действия, которые человек делает каждодневно. Заканчивается потерей зрения, галлюцинациями, замедленной речью и позже летальным исходом.
- у 45% больных наблюдается сбой когнитивных функций и мозжечковые нарушения, и только у 20% их тандем;
- симптоматика основывается на расстройстве поведения, сбое в высших корковых функциях, снижении зрения и слепоте, мозжечковой дисфункции и т. п.;
- припадки эпилептического плана: как обычные, так и при тактильном контакте;
- терминальная стадия проходит с выраженными когнитивными расстройствами;
- смерть через 7-9 месяцев после первых признаков патологии.
Передается на генном уровне, в семьях, где есть повреждение гена прионового протеина. Именно он легче всего трансформируется в патогенный белок. Имеет симптоматику и течение, аналогичное классической форме БКЯ.
Источник этой формы болезни — непреднамеренное внесение прионов пациенту при хирургическом вмешательстве, приеме некоторых лекарственных препаратов. В некоторых случаях это мозговые оболочки, которые берутся от мертвого донора для закрытия ран на мозге у живых людей.

На сегодняшний день заражение по этому источнику устранено, иногда фиксируют единичные случаи в слаборазвитых странах. Клиническая картина и течение такое же, как и классической БКЯ.
Коровье бешенство у людей по этой форме впервые диагностировали у англичан в 1995 году и пострадали от нее около 100 человек. Заражение произошло через употребление в пищу мясной продукции, в которую попали прионы бешеных животных. Другими причинами болезнь не может вызываться. Поражает она не только пожилых людей, но и молодых. У них начало недуга сопровождается серьезными изменениями в личности.
Например, они забрасывают ежедневные дела, избегают любимых и близких людей, погружаются в депрессию. Позже худеют и имеют нарушения в координации. Ближе к финалу больные не могут соблюдать гигиенические меры, кушать без чужой помощи. Все жизненные функции подавлены, и наступает летальный исход.
Важно! Модифицированные белки в этой форме БКЯ отличаются от классической болезни, т. к. слабоумие отдаленное, и человек не осознает своего состояния.
Врачи диагностируют у больных БКЯ следующие клинические проявления:
- проблемы с памятью;
- рассеянность внимания;
- поведенческие изменения;
- проблемы с координацией;
- атаксия даже на начальной стадии.
Поздние стадии характеризуются миоклоническими судорогами, особенно когда больной пугается, например, громкого звука или от другого сенсорного стимула. Для них характерны психические дисфункции от тревожности до депрессии и потери памяти.
К деменции присоединяются расстройства невралгического плана, такие как галлюцинации и/или эпилепсия и т. п. Нередко БКЯ сопровождается нарушением зрения: как частичным, так и полным.
Чтобы диагностировать этот синдром, врачи назначают диффузионно-взвешенное МРТ, маркеры ЦСЖ и некоторые другие исследования. Под обследование попадают пожилые лица, у которых бывают миоклонические судороги или атаксия. Нередко ряд расстройств может давать схожую клиническую картину, и их нужно исключить. Это может быть симптоматика:
Сложнее диагностировать патологию у молодых людей. В группе риска члены семей, где была подтверждена БКЯ новой формы или передающаяся генетически.
Вылечить болезнь Крейтцфельдта Якоба невозможно. Для всех пациентов итог одинаков — летальный исход. Однако сейчас разработаны препараты, которые замедляют поражение нейронов патогенными белками, что немного продлевает жизнь и убирает негативную симптоматику.
Прогноз для этого синдрома всегда неблагоприятный. Редко пациенты живут больше полугода от начала негативной симптоматики. Среди физических осложнений основные:
- недостаточность: как дыхательная, так и сердечная;
- инфекционные заболевания;
- патологии зрительной системы.
Несмотря на годы исследований и миллионы вложенных средств, найти лекарство от этого недуга так и не удалось. Поэтому этот диагноз всегда смертельный приговор.

Профилактики БКЯ для пациентов нет, кроме тех случаев, когда нужно следить за качеством мясной продукции. Но не стоит отказываться от консультации у генетика, чтобы выявить проблему и узнать, входит ли ваша семья в группу риска. А вот государство, врачи и люди, которые ухаживают за больными, должны принимать меры профилактики и безопасности.
При лечении и/или уходе за больным с БКЯ или коровьим бешенством нужно соблюдать ряд рекомендаций:
- тщательно вымывать руки или другие участки кожи перед едой или курением, если они касались эпидермиса зараженного или на нее попали его выделения;
- защищать лицо и конечности от попадания на них любой биологической жидкости пациента;
- при наличии ран они должны быть закрыты водостойкими повязками для недопущения выделений в организм.
Именно на руководство государства возложены основные профилактические мероприятия относительно болезни Крейтцфельдта Якоба. В нашей стране запрет на использование костной муки и мясных составляющих при производстве кормов для животных был введен в 2001 году.
А также запрещена продажа продуктов, если для их производства было использовано мясо скота, возраст которых превышает 2 года. Такие требования присутствуют в законодательствах практически всех стран мира. Именно эти мероприятия позволили избежать распространения патологии и снизили риски заражения до минимума.
В отношении генетической формы болезни никакие меры не предпринимаются. Некоторые ученые предлагали ввести принудительную стерилизацию членов семьи после подтверждения модифицированного гена в их организме, чтобы не допустить развития БКЯ.
Но по этическим соображениям это предложение не было принято даже на рассмотрение, хотя после его публикации шли жаркие споры. Ведь многие считают это гуманным, т. к. больной страдает и сам, и несет переживания своей семье, и возможности помочь им нет.
источник
При болезни Крейтцфельдта-Якоба развивается, так называемая, губчатая энцефалопатия, возникающей в результате отложения в сером веществе мозга структурно деформированных белков – прионов. Во многих случаях быстро прогрессирующее изменения приводят к смерти.
В 70-е годы было сделано открытие первых ненормальных, самовоспроизводящихся белков, полученных из тканей животных. Их назвали прионами (от английского названия белковых инфекционных частиц).
Характерной их особенностью является то, что химически (с точки зрения атомной структуры) они совпадают с правильными белками, но отличаются от них пространственной структурой. Из-за этого нерастворимы, не правильно метаболизируются и накапливаются в тканях, вызывая группу заболеваний, называемых прионными болезнями.
Считается, что их поступление в организм человека (например, с пищей) может быть причиной инфекции и привести к развитию заболевания. Вызывают дегенерация периферической нервной системы (энцефалопатию). Наиболее распространенным заболеванием этой группы является болезнь Крейтцфельдта-Якоба.
Болезнь Крейтцфельдта-Якоба – это прионная болезнь, приводящая к губчатому поражению головного мозга. Она влияет на серое вещество и являются причиной постепенного исчезновения нервных клеток – нейронов.
Группа симптомов, возникающих в результате этих изменений:
- во время начальной стадии – гиперактивность, агрессия, расстройства настроения, потеря аппетита
- быстро прогрессирующая деменция
- непроизвольные, повторяющиеся мышечные сокращения отдельных мышц или группы мышц, то есть миоклонус
- изменения в электроэнцефалограмме – т.е. записи электрической активности мозга
Картина ЭЭГ при болезни Крейтцфельдта-Якоба очень характерна, но этот симптом встречается не у всех больных. Довольно часто появляются и другие неврологические симптомы, такие как нарушение координации движений, нарушение походки. Больной может иметь нарушения ритма бодрствования и отдыха, страдать от бессонницы, иметь проблемы с сосредоточением внимания на самых простых действиях или разговоре. Присоединяются к этой картине нарушения функционирования некоторых черепно-мозговых нервов, что вызывает нарушения речи или подвижности глазных яблок.
Развивающаяся деменция вызывает:
- изменчивость настроения
- раздражительность, иногда агрессивность
- нарушения памяти (особенно оперативной)
- проблемы с ориентацией в отношении времени, места, идентификации собственной личности и близких
- спутанность сознания
- галлюцинации и бред
Этот не очень характерный набор симптомов, поэтому необходимо дифференцирование болезни Крейтцфельдта-Якоба с болезнями Альцгеймера, Паркинсона и другими нейродегенеративными заболеваниями.
Аналогичные заболевание диагностируется также у крупного рогатого скота. В просторечии их называют «коровье бешенство».
Болезнь Крейтцфельдта-Якоба встречается в четырех основных описанных формах. Наиболее распространенной является спорадическая форма, причины которой установить не удаётся. Не обнаруживается корреляции с возможными внешними или внутренними причинами. Вызывает прогрессирующую деменцию, нарушение равновесия, появление движений, независимых от воли больного, исчезновение чувствительности поверхностного и глубокого типа (на одной стороне тела).
Второй формой является семейный тип заболевания Крейтцфельдта-Якоба (fCJD) – в этом случае было установлено, что причиной заболевания является мутация генов белков, в результате чего образуются прионы. Больные страдают от мозговых симптомов – характерные нарушения походки, речи, нистагм, тремор движения. Тип fCJD составляет 10-15% всех случаев заболевания.
Ятрогенная форма болезни Крейтцфельдта-Якоба (iCJD) возникает в результате поступления инфекционного (содержащего прионы) материала во время медицинских процедур или лечения препаратами животного происхождения. Этот тип заболевания обусловил значительное ограничение использования инструментов многоразового использования, так как стандартный процесс стерилизации не удаляет прионов.
Последней формой является вариативная болезнь Крейтцфельдта-Якоба (vCJD). Считается, что причиной является употребление говядины, полученной от больных коров. Чаще всего страдают люди в возрасте 20-35 лет – что быстро вызывает глубокое ослабление разума в молодом возрасте, с большим количеством психотических симптомов. Существование этого механизма инфекции привело к тому, что крупный рогатый скот подвергается очень тщательной проверке.
Губчатая энцефалопатия (то есть, дисфункция головного мозга, вызванная прионами) является прогрессирующим и необратимым состоянием. В настоящее время нет эффективных методов её лечения. Лечебные действия направлены на замедление развития болезни и продление поддержания работоспособности больного, но прогноз всегда пессимистичный.
источник

Прионные заболевания – это особый вид тяжелых нейродегенеративных болезней человека и животных, которые вызываются особыми возбудителями: прионами. Основные сведения о прионных болезнях нервной системы, то есть: что собою представляют прионы, как они вызывают поражение нервной системы, какие заболевания вызывают и как с ними бороться, — вы сможете узнать из этой статьи.

Прионы – это белковые структуры. Эти белковые молекулы могут быть нормальными, входящими в состав тканей человека и животных, то есть содержащимися в организме здоровых людей и других млекопитающих, а могут быть патологическими, вызывающими заболевания.
Ранее было принято считать, что любая «живая» структура обязательно содержит нуклеиновые кислоты (ДНК, РНК), которые позволяют ей размножаться. Все вирусы, бактерии, грибы, птицы, животные, люди содержат нуклеиновые кислоты. И предполагалось, что отсутствие этих кислот означает невозможность размножения, однако открытие прионов перевернуло эти представления.
Прионы состоят только из белка, но при этом умеют размножаться. Попадая в организм, они вызывают превращение содержащегося в нем нормального приона в патологический, таким способом увеличивая свое количество. То есть все случаи столкновения с нормальными прионами организма заканчиваются образованием патологических прионов. На это требуется значительно большее количество времени, чем при размножении вирусов или бактерий, поэтому от момента попадания приона в организм до развития заболевания иногда проходят годы.
Прионы были открыты в 1982 году Стэнли Прузинером, за что в 1997 году он был удостоен Нобелевской премии.
До открытия прионов был описан ряд заболеваний человека и животных, причина которых никак не могла быть установлена. Еще в XVIII веке в Великобритании была зарегистрирована так называемая «почесуха» овец с симптомами в виде сильного зуда, нарушений движений в конечностях и припадков, которые свидетельствовали о поражении нервной системы. Так, в 1957 году американец Карлтон Гайдушек описал заболевание у племени Фор, обитающего на высокогорьях Папуа-Новой Гвинеи. Заболевание было связано с каннибализмом и передавалось, таким образом, от одного человека к другому. С 1986 года в Англии, а затем и в других странах было зарегистрировано заболевание крупного рогатого скота, названное «коровьим бешенством» (хотя к возбудителю бешенства оно не имело никакого отношения). «Коровье бешенство» приобрело масштабы эпидемии и вызывалось прионами. В 90-е годы была доказана передача «коровьего бешенства» человеку вместе с употребляемым мясом больного животного. На сегодняшний день изучение ряда заболеваний с неустановленными причинами привело к тому, что высказана гипотеза о прионной природе этих болезней (например, болезнь Альцгеймера, болезнь Паркинсона и другие). Несмотря на успехи в этом направлении, еще очень многое остается за гранью постигнутого.
Прионы – это очень устойчивые соединения. Большинство способов обезвреживания и обеззараживания оказываются неэффективными при борьбе с ними. Они не погибают при кипячении в течение 2-3 часов, выдерживают холод до -40°С несколько лет, не чувствительны к ультрафиолетовому излучению и радиации, не инактивируются при обработке формалином.
Особое строение прионов приводит к тому, что организм не умеет бороться с ними. Человеческий организм не вырабатывает антитела (гуморальный иммунитет) против них, не атакует прионы лимфоцитами (клеточный иммунитет), как бы не замечает их. То есть попадание прионов в организм неизбежно влечет за собой развитие заболевания. Даже при 10-миллионном разведении прион оказывается заразным для человека.

На сегодняшний день известны три способа заражения:
- трансмиссивный: при передаче приона от одного вида млекопитающего к другому. Причем если ранее говорилось о существовании межвидовых барьеров, то есть передача от коровы или овцы к человеку отрицалась, то на сегодняшний день считается, что прионы могут передаваться от любого инфицированного животного или человека (теоретически), и доказана возможность заражения человека от коров и другого человека. Причиной попадания патологического приона может быть употребление в пищу мяса больного животного, использование биологических тканей животных и человека (препараты крови, пересадка роговицы или твердой мозговой оболочки и тому подобное). Считается, что различные биоматериалы обладают разной степенью патогенности. Так, наиболее заразными являются ткани мозга и мозговых оболочек ввиду наибольшего содержания в них прионов, затем – кровь и ее препараты. Попадание возбудителя под кожу и через рот (с пищей) относят к наименее заразным способам. Это означает определенную дозозависимость прионных заболеваний;
- наследственный: когда заболевание развивается в результате генной мутации, возникающей в определенной области 20-й хромосомы. Особый участок 20-й хромосомы человека отвечает за наличие в организме нормального прионного белка. Функции его до конца так и не изучены. При возникновении генных нарушений вместо нормального приона синтезируется патологический, вызывающий заболевание;
- спорадический: спонтанное возникновение аномального белка в организме человека.
Таким образом, становится понятно, что прионные заболевания могут быть как инфекционными, так и наследственными. Каким бы путем прион не возник в организме человека, он может стать причиной заражения другого человека. Это означает, что даже случайным образом возникшая прионная болезнь может передаться другому человеку трансмиссивно. Предполагается различная степень инфекционности прионов, возникших в результате мутации или спонтанно, а также полученных от другого вида млекопитающих.
Прионы способны вызывать так называемые губчатые (спонгиозные) энцефалопатии, то есть поражение центральной нервной системы. Морфологически это означает:
- образование в клетках мозга полостей («дырок», отчего и сформировалось понятие «губчатости»);
- гибель (дегенерацию) нейронов;
- разрастание соединительной ткани вместо погибших нервных клеток;
- атрофию мозга;
- формирование амилоидных бляшек (последние образуются из скоплений патологических прионов).
И все это возникает на фоне полного отсутствия каких-либо воспалительных реакций.
На сегодняшний день точно известно несколько заболеваний человека, виною которых являются прионы:
- болезнь Крейтцфельда-Якоба;
- болезнь Герстманна-Штреусслера-Шейнкера;
- болезнь куру;
- семейная фатальная инсомния (бессонница);
- хроническая прогрессирующая спонгиозная энцефалопатия детского возраста (болезнь Альпера).
Эта разновидность прионной инфекции довольно многообразна, потому что в настоящее время принято выделять несколько ее форм:
- спорадическую (классическую);
- семейную (наследственную);
- ятрогенную;
- новую атипичную форму, возникающую в результате инфицирования мясом крупного рогатого скота, пораженного «бешенством коров», то есть эта форма представляет собой аналог «коровьего бешенства» у людей.
Спорадическая форма с момента описания этого заболевания в 20-х годах прошлого столетия была наиболее часто встречающейся (около 85-90% всех случаев болезни Крейтцфельда-Якоба). Она возникает преимущественно в пожилом возрасте, первые признаки появляются после 55 — 60 лет. Однако в последние годы статистика изменилась после возникновения эпидемии «коровьего бешенства». Все чаще и чаще стали регистрироваться случаи новой атипичной формы в результате заражения от коров. Для этой формы характерно более раннее появление: заболевают молодые люди. Заболеваемость спорадической формой составляет 1 случай на 1 миллион населения в год. Характерно постепенное появление различных симптомов, которые делятся на две группы: неврологические и психические. Появление тех или иных изменений обусловливается областью поражения в головном мозге. Начальными симптомами могут быть: головная боль, головокружение, нарушение сна, снижение полового влечения, расстройство аппетита, общая слабость и повышенная утомляемость. Постепенно развиваются ухудшение памяти и внимания, нарушение мышления, потеря зрения, депрессия, эмоциональная лабильность. Психические нарушения могут протекать и довольно остро в виде эпизодов дезориентации, галлюцинаций и бреда. Из неврологических симптомов формируется мышечная слабость во всех конечностях (парезы), нарастает мышечный тонус, нарушается речь вплоть до полной утраты, возникает выраженная координаторная неустойчивость (атаксия), могут появляться непроизвольные движения в конечностях (особенно характерны миоклонии: быстрые неритмичные малоамплитудные сокращения небольших мышц), а также судорожный синдром. Болезнь неуклонно прогрессирует и в конечной стадии приводит к обездвиженности, утрате контроля над функцией тазовых органов, деменции и кахексии. Продолжительность жизни с момента появления первых симптомов составляет не более 2-х лет. Исход заболевания – смерть в 100% случаев.
Семейная форма связана с генными мутациями в области 20-й хромосомы. Составляет около 5-6% всех случаев болезни Крейтцфельда-Якоба. Заболевание носит аутосомно-доминантный характер, что означает, что оно не связано с полом, а также то, что патологический ген проявляет себя всегда, независимо от того, нормален ли второй аналогичный ген (все гены человека – парные). Первые признаки заболевания возникают приблизительно на 5-10 лет раньше, чем при спорадической форме. В остальном течение заболевания сходно с предыдущей формой.
Ятрогенная форма возникает в результате заражения человека во время медицинских вмешательств. Статистика этой формы заболевания отсутствует, поскольку довольно сложно проследить и доказать именно такой путь заражения. Существуют подтвержденные документально случаи заражения при использовании твердой мозговой оболочки, роговицы, стереотаксических операциях на головном мозге (в основном, эти случаи относятся к Франции и Австралии). Инкубационный период (время от заражения прионами до появления первых признаков заболевания) колеблется от 7 месяцев до 12 лет. Длительность инкубационного периода зависит от нескольких факторов: места и способа проникновения прионов в организм, дозы возбудителя, исходного генотипа организма человека. Быстрее всего болезнь развивается при непосредственном попадании прионов в ткань мозга при проведении оперативных вмешательств, чуть более длительный инкубационный период при инфицировании в результате пересадки твердой мозговой оболочки и роговицы. Попадание прионов в организм при введении инфицированных препаратов (например, человеческого гонадотропина) сопровождается развитием заболевания через 5-10 лет. Для этой формы заболевания характерно преобладание неврологических симптомов над психическими. Также развиваются выраженная мозжечковая атаксия, нарушения речи, изменения мышечного тонуса, миоклонии. Неизменно развивается деменция. Больные погибают в течение одного — максимум 2-х лет.
«Коровье бешенство» у людей, или новая атипичная форма болезни Крейтцфельда-Якоба стала приобретать актуальность после эпидемии прионной инфекции у крупного рогатого скота в Англии в 90-х годах. Употребление в пищу мяса пораженных животных стало причиной инфицирования прионами. Для этой формы заболевания характерно гораздо более раннее начало: регистрируются случаи и в 30, и в 40 лет. Как и при ятрогенной форме, неврологические проявления преобладают над психическими. Однако на прогноз это не влияет. Эта форма заболевания также смертельна для человека.
Этот вариант прионного заболевания относится к наследственному. Встречается довольно редко: 1 случай на 10 000 000 населения. Передается по аутосомно-доминантному типу. Обычно заболевание проявляет себя в возрасте пациента около 40 лет. Первыми и доминирующими в клинической картине являются мозжечковые расстройства. Вначале появляется головокружение и шаткость при ходьбе. Координаторные нарушения прогрессируют, произвольные движения становятся избыточными и «промахивающимися», постепенно самостоятельная ходьба становится невозможной. Наряду с этими симптомами возникают нарушения мышечного тонуса, снижение слуха вплоть до глухоты, снижение зрения до слепоты, парез взора вверх (когда человек не может посмотреть вверх одновременно двумя глазами), проблемы с глотанием и звуковоспроизведением. Деменция появляется в терминальной стадии заболевания. Средняя продолжительность жизни составляет от 2-х до 10 лет. Исход заболевания – смерть.
Эта разновидность прионной инфекции изучена благодаря племени людоедов, живущих на высокогорьях Папуа-Новой Гвинеи. До 1956 года среди населения этой географической зоны были распространены традиции ритуального каннибализма: поедание мозга умерших родственников. Предполагается, что у одного из членов племени спорадически возникла прионная инфекция, которая потом распространилась из-за съедения его мозга соплеменниками. После отмены традиций каннибализма заболевание стало регистрироваться значительно реже, на сегодняшний день практически не встречается. Инфекционное начало болезни и заразность были доказаны Гайдушеком путем заражения шимпанзе вытяжкой из мозга больных людей.
«Куру» на языке этого племени означает «смеющийся» или «дрожащий от страха». Такое название не случайно. После инкубационного периода длительностью от 5 до 30 лет появляются мозжечковые расстройства вместе с дрожанием отдельных частей тела и неконтролируемым смехом, смазанной речью, нарушениями движений глазами, дисфункцией глотания, мышечной слабостью. Мышление вначале не нарушается, деменция развивается уже в терминальную стадию. Больные живут от 4 до 36 месяцев с момента появления первых симптомов.

Это прионное заболевание известно науке с 1986 года. Очень редко встречается. Относится к наследственным заболеваниям с аутосомно-доминантным типом передачи. К 2003 году было описано 26 семей с семейной фатальной инсомнией. Имеет довольно вариабельный возраст начала заболевания: от 25 лет до 71 года. Продолжительность жизни с момента появления первых признаков составляет от 6 до 48 месяцев.
Самым главным признаком заболевания становится бессонница. Человеческий организм утрачивает способность регулировать циклы бодрствования и отдыха, вообще не может спать. Помимо бессонницы возникают двигательные нарушения в виде миоклоний, дрожания, мышечной слабости. Характерны вегетативные расстройства, проявляющиеся повышением артериального давления, повышением температуры тела, потливостью, учащенным сердцебиением. Из психических нарушений возможны зрительные галлюцинации, панические страхи, эпизоды спутанности сознания. На фоне полного отсутствия сна наступает истощение и больной погибает.
Эта разновидность прионных заболеваний развивается у детей (до 18 лет). Характеризуется передачей по наследству по аутосомно-рецессивному типу (не связана с полом и проявляется только в случае совпадения двух патологических генов от отца и от матери). Поражение нервной системы заключается в нарушении зрения, развитии эпилептических припадков. Возможны периодические острые состояния, протекающие по типу инсультов. Кроме поражения нервной системы, это заболевание сопровождается и поражением печени. Довольно быстро развивается хронический гепатит, который переходит в цирроз. Больные погибают от интоксикации из-за выраженной печеночной недостаточности в течение 12 месяцев от начала заболевания.
К сожалению, в настоящее время все прионовые болезни не излечимы. Больные получают симптоматическую терапию (например, противосудорожные препараты при эпилептических приступах), которая может только облегчить страдания, но никаким образом не влияет на прогноз. А он весьма неутешителен: все известные в настоящее время прионные инфекции смертельны для человека.
От развития спонтанных и наследственных форм прионовых инфекций обезопасить себя нельзя. Теоретически некоторые наследственные формы можно исключить (имеется ввиду определить, имеется ли подобное генетическое нарушение у конкретного человека), пройдя генетическую диагностику. Однако осуществить это весьма затруднительно, поскольку лаборатории, осуществляющие подобные анализы, можно найти преимущественно за рубежом.
Для того, чтобы хоть как-то обезопасить себя от болезни Крейтцфельда-Якоба, в первую очередь, необходимо не употреблять в пищу мясо и мясные продукты из регионов, где зарегистрированы случаи заболевания крупного рогатого скота (в основном, это страны Европы. В России действует запрет на ввоз подобной продукции из «опасных» регионов). Также не следует принимать лекарственные препараты, изготовленные из крови человека и животных (необходима замена на синтетические аналоги), по возможности избегать переливания крови.
Таким образом, прионные заболевания – это недостаточно изученные формы инфекционных и наследственных нарушений, возникающих в организме человека в результате появления патологических прионов. В большинстве случаев прионные заболевания поражают нервную систему. Результат заболевания всегда один: смерть. Пока современная медицина не располагает эффективными средствами борьбы с этими заболеваниями.
Телеканал «Россия-24», научно-познавательная передача «Прионы — маленькие убийцы». Рассказывает Адриано Агуцци — директор Швейцарского института невропатологии, занимающийся исследованием прионов.
источник