Опубликовано в журнале:
«ЖУРНАЛ НЕВРОЛОГИИ И ПСИХИАТРИИ» 2007, № 11, с. 45-48
В.В. Захаров
Кафедра нервных болезней Московской медицинской академии им. И.М. Сеченова
Одной из наиболее хорошо зарекомендовавших себя стратегий терапии когнитивных нарушений является воздействие на церебральные нейротрасмиттерные системы. Применение ацетилхолинергических и глютаматергических препаратов является в настоящее время «золотым стандартом» лечения наиболее распространенных форм деменции [1—4, 8, 20, 22], хотя применение первых на стадии умеренных когнитивных нарушений пока дало противоречивые результаты [19—21]. Предполагают, что развитие ацетилхолинергической недостаточности является относительно поздним событием в патогенезе когнитивных расстройств и менее тяжелые нарушения в большей степени обусловлены дисфункцией со стороны других нейротрансмиттерных систем — дофаминергической, норадренергической, серотонинергической и др.
В связи с этим весьма интересны результаты исследований L. Backman и соавт., N. Volkow и соавт. [15, 30]. В цитируемых работах было показано, что в пожилом возрасте закономерно развивается легкая дофаминергическая недостаточность, о чем свидетельствует уменьшение биодоступности D2-рецепторов полосатых тел (по данным позитронно-эмиссионной томографии головного мозга). При этом прослеживается корреляция между указанными нейрохимическими изменениями и выраженностью когнитивных нарушений лобного типа, которые являются одним из основных и наиболее ранних признаков хронической сосудистой мозговой недостаточности [2, 4, 7, 12].
В основе данной корреляции вероятно лежит недостаточность дофаминергической активации лобных долей со стороны вентральной зоны покрышки среднего мозга (так называемый мезокортикальный дофаминергический путь).
К настоящему времени накоплен значительный клинический опыт применения дофаминергических препаратов при возрастной когнитивной дисфункции. Наиболее изученным препаратом является пирибедил, или проноран (фирма «Сервье»). Он совмещает свойства агониста дофаминовых D2/DЗ-рецепторов и блокатора пресинаптических а2-адренорецепторов. Применение пронорана способствует усилению не только дофаминергической, но и норадренергической медиации, что весьма выгодно с клинической точки зрения, так как активация норадренергической системы играет одну из ключевых ролей в процессе запоминания новой информации. Кроме того, по экспериментальным данным проноран обладает также положительным вазоактивным эффектом в отношении церебрального и периферического кровообращения, что также является преимуществом при использовании данного препарата у пациентов с хронической сосудистой мозговой недостаточностью [5, 6, 14, 15, 24, 27, 28].
В 70—80-х годах XX века в европейских странах проводилась серия плацебо-контролируемых клинических испытаний, в которых был показан положительный ноотропный эффект пронорана у пациентов с возрастной когнитивной дисфункцией, не достигающей выраженности деменции [14, 15, 27, 28]. При этом, по некоторым данным, эффективность пронорана превосходила эффективность монотерапии сосудистыми препаратами [28]. В 2001 г. эти результаты были воспроизведены в работе D. Nagaraia и соавт. [25], которые применяли проноран у пациентов с синдромом умеренных когнитивных нарушений. Было показано, что применение пронорана сопровождалось достоверно большим процентом улучшений когнитивных функций по Краткой шкале оценки психического статуса по сравнению с плацебо. Известно также, что на ранних стадиях болезни Паркинсона использование пронорана сопровождается не только улучшением двигательных, но и когнитивных функций [10].
Согласно требованиям доказательной медицины, проведение двойных слепых плацебо-контролируемых рандомизированных исследований является основным и абсолютно необходимым методом оценки эффективности ноотропных препаратов. Однако этот метод также имеет определенные ограничения, которые препятствуют «механическому» перенесению полученных результатов на повседневную клиническую практику. Как известно, отбор пациентов при проведении клинических исследований проводится по определенным критериям, которые, как правило, значительно ограничивают число сопутствующих заболеваний и применение прочих препаратов. Также не всегда просто интерпретировать практическое значение полученных в ходе рандомизированных исследований изменений психометрических шкал. В связи с этим целесообразен дополнительный анализ результатов применения того или иного препарата в условиях обычной клинической практики. При этом наряду с психометрическими [например «Краткой шкалой оценки психического состояния» (Mini-mental scale examination — MMSE)] используются шкалы типа шкалы «Общего клинического впечатления» (Clinical Global Impression — CGI). Последние предоставляют лечащему врачу значительную свободу в оценке практического значения проводимой терапии на основании всей совокупности изменений клинического статуса пациента. Применение таких шкал является общепринятым в мировой практике и настоятельно рекомендуется [23, 26] для применения в области когнитивной неврологии.
В 2005—2007 г. в России в рамках программы «Прометей» (координатор проекта — акад. РАМН Н.Н. Яхно) был проанализирован опыт применения пронорана в терапии возрастной когнитивной дисфункции в повседневной клинической практике. Результаты выполнения этой программы были опубликованы в целом ряде статей [6, 9, 14].
Данная публикация отражает раздел указанной программы, целью которого была оценка влияния препарата проноран на возрастные когнитивные нарушения при определении их выраженности разными методами. В этом случае сопоставляли данные психометрических шкал (MMSE) [18] и нейропсихологических тестов (тест рисования часов) [23], с одной стороны, и клинической шкалы (CGI) [27], с другой.
Материал и методы
В соответствии с указанной целью исследование было разделено на 2 части. В 1-й части для оценки эффективности терапии использовались психометрические (MMSE) и нейропсихологические тесты, а во 2-й части — шкала CGI.
В 1-й части был проведен анализ лечения 574 пациентов в возрасте от 60 до 89 лет (в среднем 69,5±5,5 года) с легкими или умеренными когнитивными нарушениями возрастного и/или сосудистого характера. Эти пациенты наблюдались в поликлиниках 33 городов из 30 регионов России. Наиболее частым клиническим диагнозом была дисциркуляторная энцефалопатия I или II стадии. В лечении и наблюдении за пациентами приняли участие 132 врача-невролога амбулаторного звена.
Проноран назначали в дозе 50 мг в сутки в 1 прием в течение 3 мес. Монотерапия данным препаратом проводилась 385 пациентам, а 189 пациентов получали проноран в сочетании с другими сосудистыми и метаболическими средствами. Оценка эффективности терапии проводилась на 6-й и на 12-й неделе лечения.
Во 2-й части был проведен анализ лечения 2058 пациентов (1447 женщин и 611 мужчин в возрасте от 50 до 94 лет, в среднем 64,9+8,3 года) с диагнозом дисциркуляторная энцефалопатия I или II стадии и легкими или умеренными когнитивными нарушениями. Данные пациенты наблюдались в поликлиниках 54 городов из 37 регионов России. В лечении и наблюдении за пациентами приняли участие 212 врачей-неврологов амбулаторного звена.
В этой группе проноран пациентам назначали в дозе 50 мг в сутки в 1 прием в течение 3 мес. Монотерапия этим препаратом проводилась 1181 пациенту, а 875 получали проноран в сочетании с другими сосудистыми и метаболическими средствами.
Оценка эффективности терапии в этих случаях также проводилась на 6-й и на 12-й неделе лечения. Различий по возрасту, полу и выраженности когнитивных расстройств между пациентами, получавшими мототерапию пронораном и политерапию в обеих группах не было.
По CGI лечащий врач до начала терапии, и далее на 6-й и 12-й дни оценивал общее состояние пациента на основании всей совокупности имеющейся симптоматики по 7-балльной шкале оценке «нет нарушений» соответствовал 1 балл, «незначительные» — 2, «легкие» — 3, «умеренные» — 4, «умеренно тяжелые» — 5, «тяжелые» — 6, «крайне тяжелые нарушения» — 7 (эти баллы далее см. на рисунке).
Кроме того, отмечалась степень влияния соответствующих нарушений на повседневную жизнь больных.
Как в 1-й, так и во 2-й частях исследования в анализ не включались пациенты в остром периоде инсульта, черепно-мозговой травмы, после оперативного вмешательства, с болезнью Паркинсона и синдромом паркинсонизма, с шизофренией, а также пациенты с тяжелыми речевыми, двигательными и чувствительными расстройствами.
Результаты и обсуждение
На фоне терапии пронораном было отмечено достоверное улучшение когнитивных функций. Так, средний результат по MMSE повысился с 26,0±1,0 до лечения до 27,9±3,5 балла на 12-й неделе лечения (pРаспределение больных (в %) по показателям шкалы CGI до лечения, на 6-й и 12-й неделе терапии.
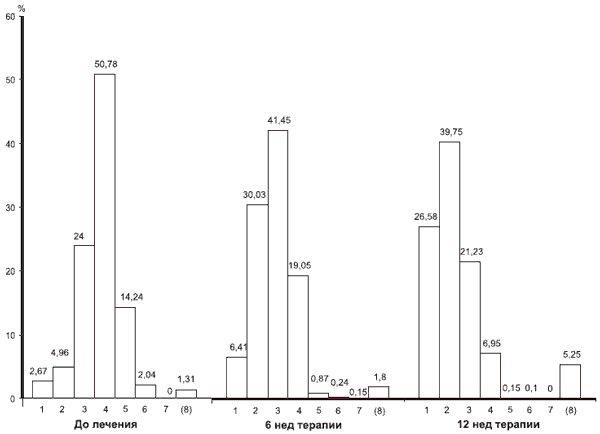
По оси ординат — процент больных, по оси абсцисс — баллы (1—7); (8) — нет данных.
На фоне проводимой терапии достоверно уменьшилась также доля пациентов, допустивших существенные ошибки в тесте рисования часов. До лечения такие ошибки выявлялись у 48,6% пациентов, после лечения (на 12-й неделе) — лишь у 14,3% (p Улучшение когнитивного функционирования больных было отмечено и по шкале CGI. Если до лечения выраженность нарушений по данной шкале у большинства пациентов варьировала от легкой до умеренно тяжелой (3—5 баллов), то после лечения это были или умеренные либо незначительные (2—4 балла) расстройства. Отсутствие динамики зафиксировано у 170 пациентов, а отрицательная — только у 6. Распределение больных в соответствии с показателями шкалы CGI до лечения, на 6-й и 12-й неделе терапии приведено на рисунке.
Терапия пронораном характеризовалась удовлетворительным профилем безопасности и переносимости. Нежелательные явления отмечались у 15,4% пациентов. Наиболее распространенными из них были тошнота, головокружение, рвота, головная боль, повышенная сонливость, боли или дискомфорт в эпигастрии, диспепсия, общая слабость. Данные явления не угрожали жизни пациентов, не наносили непоправимого вреда здоровью и крайне редко (менее 1% случаев) вынуждали прекратить проводимую терапию. Несколько чаще нежелательные явления отмечались у пациентов, получавших комбинированную терапию, по сравнению с получавшими монотерапию пронораном (15 и 22% соответственно).
Таким образом, опыт практического применения пронорана в условиях повседневной амбулаторной неврологической практики согласуется с результатами опубликованных ранее плацебо-контролируемых рандомизированных клинических исследований.
Применение пронорана у пациентов с хронической сосудистой мозговой недостаточностью способствует регрессу выраженности когнитивных нарушений. При этом улучшение когнитивных функций фиксируется не только с помощью психометрических, но общих клинических шкал. Данный факт свидетельствует о практической значимости выявленного когнитивного улучшения, которое, по нашим наблюдениям, оказывает ощутимое положительное влияние на повседневную жизнь пациента, его поведение и самочувствие.
Сказанное дает основание утверждать, что терапия пронораном показана для лечения пациентов с недементными (легкими или умеренными) когнитивными нарушениями. Это имеет практическое значение, ибо на сегодняшний день отсутствует единый протокол ведения пациентов с когнитивными расстройствами, не достигающими стадии деменции [7, 9, 11, 12, 20], хотя распространенность недементных нарушений в пожилом возрасте значительно превышает частоту деменции [6, 16, 18]. При этом они, снижая качество жизни не только пациентов и их ближайших родственников, приносят социально-экономический ущерб обществу в целом. Поэтому раннее начало лечения когнитивных нарушений имеет большое значение и для профилактики деменции, так как пациенты с умеренными когнитивными нарушениями, представляют собой группу высокого риска ее развития в будущем (так, заболеваемость болезнью Альцгеймера в данной категории пациентов составляет 10-15% в год [25]).
Из изложенного видно, что дофаминергическая терапия может проводиться как самостоятельный вид лечения недементных когнитивных нарушений, так и в комплексе с традиционными сосудистыми и ноотропными препаратами. Важно, что проноран может сочетаться с другими ноотропными и сосудистыми препаратами, поскольку такое сочетание не приводит к увеличению частоты побочных эффектов. Вместе с тем следует обратить внимание на то, что в нашей работе не было получено различий по величине ноотропного эффекта между монотерапией пронораном и комбинацией данного препарата с другими сосудистыми и ноотропными лекарственными средствами. Это дает нам основание сделать вывод, что указанная комбинированная терапия не имеет в данном случае особых преимуществ в отношении коррекции когнитивных расстройств по сравнению с монотерапией пронораном.
Таким образом, проноран оказывает положительный ноотропный эффект при недементных (легких и умеренных) когнитивных нарушениях сосудистой этиологии. Данный эффект не фиксируется с помощью различных методов оценки нейропсихологических тестов и клинически ориентированных шкал, позволяющих оценить состояние больных в целом.
Эффективность в отношении когнитивных расстройств и удовлетворительный профиль переносимости и безопасности у пациентов пожилого возраста позволяет рекомендовать проноран для широкого клинического применения при дисциркуляторной энцефалопатии I и II стадии.
источник
ФУНКЦИЯ ИММУННОЙ СИСТЕМЫ ПРИ СТАРЕНИИ И БОЛЕЗНИ АЛЬЦГЕЙМЕРА
Г. И. Коляскина, Л. В Андросова, Т. П. Секирина, С. Г. Кушнер, О. А. Бурбаева,
Е. Ф. Васильева, С. И. Гаврилова, Н. Д. Селезнева Научный центр психического здоровья РАМН, Москва
Иммунная система и старение. В литературе имеются сведения о том, что возрастные изменения иммунной системы начинаются довольно рано — в 130-летнем возрасте [2]. Они прежде всего касаются субпопуляции тимусзависимых лимфоцитов (Т-лимфоцитов), которые составляют 70 % всех лимфоцитов иммунной системы. Такие сдвиги рассматриваются как следствие эволюции тимуса (вилочковой железы), которая начинается в этом возрасте. В дальнейшем по мере старения изменения функции иммунной системы проявляются в снижении гуморального и клеточного иммунного ответа на чужеродные антигены, увеличении частоты возникновения и выраженности клеточных и гуморальных аутоиммунных реакций, уменьшении способности к развитию иммунологической толерантности, повышении уровня циркулирующих иммунных комплексов и ряде других сдвигов [1,16]. Некоторые авторы [1,2,7,9] отмечают также уменьшение общего числа лимфоцитов в периферической крови, особенно Т-лимфоцитов и изменение соотношения Т-лимфоцитов-хелперов и Т-лимфоцитов-супрессоров, снижение способности лимфоцитов к пролиферации при их стимуляции антигенами, митогенами и т. п.
По нашим данным, у здоровых людей в возрасте от 48 до 67 лет по сравнению с людьми 18-30 лет общее количество лимфоцитов достоверно не изменяется, составляя 1,8х10 6 клеток в 1 мл. При этом число лимфоцитов одинаково у мужчин и женщин пожилого возраста — 1,84хЮ 6 и 1,76хЮ 6 соответственно. Нами не обнаружено также статистически достоверных изменений числа Т-популяций лимфоцитов и соотношения субпопуляций Т-лимфоцитов-хелперов и Т-лимфоцитов-супрессоров у лиц 48-70 лет по сравнению с обследованными 18-30 лет, наблюдалась лишь тенденция к понижению числа Т-лимфоцитов у старых людей по сравнению с молодыми (54,0±6,0 и 74,0±5,0 % соответственно), к снижению количества Т-лимфоцитов с хелперной активностью (54,0±0,4 и 43,0±5,0 %) и повышению приблизительно на 20 % количества Т-лимфоцитов с супрессорной активностью (32,0±2,0 и 40,0±5,0 %), чему соответствовало и снижение соотношения Т-хелперов и Т-супрессоров в пожилом возрасте (с 1,7 до 1,1). Число лимфоцитов, зависимых от функции костного мозга (В-лимфоцитов), с возрастом увеличивалось на 75 %.
Процесс нормального старения сопровождается значительным снижением как уровня пролиферативной активности Т-клеток в ответ на стимуляцию митогенами, так и уровня продукций интерлейкина -1 (ИЛ-1) и интерлейкина-2 (ИЛ-2) лимфоцитами крови.
Изменений функциональной активности В-лимфоцитов у людей пожилого возраста по сравнению с молодыми людьми обнаружено не было.
Наши данные свидетельствуют и о том, что старение сопровождается статистически достоверным (р в возрасте после 48 лет не изменяются (74,0±4,0 — у пожилых в возрасте от 48 до 70 лет и 76,0±10,0 — у лиц старше 70 лет).
Из литературы известно, что проблема возрастных изменений иммунной системы не может быть рассмотрена без учета иммунореактивности, определяющей резистентность к инфекционным болезням и к опухолевому росту [8]. Иммунореактивность в большой мере связана с функцией лимфоцитов — естественных киллеров (ЕК), активность которых по данным литературы изменяется с возрастом [5,7]. Нами также установлены факты, свидетельствующие о снижении функциональной активности ЕК у здоровых лиц пожилого возраста (старше 40 лет) по сравнению с таковой у лиц молодого (до 30 лет) возраста (29,0±8,0 и 50,5±5,0 % соответственно; р
Поскольку число Т-лимфоцитов в процессе старения уменьшается незначительно, то этими сдвигами вряд ли можно объяснить снижение их функциональной активности. В связи с этим могут представлять интерес данные R . К. Chopra и соавт. [13], которые показали, что стимулированные ФГА лимфоциты крови здоровых людей молодого возраста (до 30 лет) пролиферируют приблизительно в 2 раза активнее и синтезируют в 2 раза больше биологического активного ИЛ-1, чем лимфоциты пожилых лиц. Это дает основание предположить, что сниженная пролиферативная активность Т-лимфоцитов в пожилом возрасте определяется недостатком ИЛ-2. Однако добавление даже избыточного количества ИЛ-2 в культуру Т-лимфоцитов пожилых людей не восстанавливает полностью их пролиферативную активность. Поэтому должны существовать дополнительные факторы, обеспечивающие последнюю. Исследования последних лет [26] показали, что в популяции Т-лимфоцитов пожилых людей значительную часть (50 % или больше) составляют клетки, которые не могут активироваться под влиянием митогенов и что существенную роль в способности клетки пролиферировать играет экспрессия рецепторов для ИЛ-2 на ее поверхности. По-видимому, значительное количество Т-клеток старых людей из-за сниженной экспрессии указанных рецепторов и недостатка ИЛ-2 неспособно пройти весь цикл активации. При этом, поскольку отдельные Т-лимфоциты в отношении способности их рецепторов взаимодействовать с лимфокинами (ИЛ-2), могут изменяться в разной степени [25], то снижение функциональной активности Т-лимфоцитов при старении происходит за счет накопления в крови не реагирующих соответствующим образом Т-лимфоцитов. К этому следует добавить и возможность их гибели в процессе активации.
Несмотря на отмеченные особенности, в процессе старения организма значительного дефицита Т-лимфоцитов не возникает. Снижается лишь приток новых, частично дифференцированных клеток, что связывают с торможением активности тимуса в процессе его инволюции и утратой части предшественников иммунокомпетентных клеток. Есть также мнение, что определенную роль в развитии изменений в иммунной системе в процессе старения играют нарушения в механизмах нейроиммуномодуляции [17], в частности осуществляемой разными звеньями дофаминергической системы мозга [3,4,21,27].
Болезнь Альцгеймера (БА). В последнее время в литературе привлекается внимание к иммунным нарушениям при БА [11,18,19,20,22,23], которые могут быть вовлечены в патогенез заболевания.
В начале изучения функции иммунной системы при БА возникало закономерное предположение, что в этом случае может иметь место углубление изменений, характеризующих нормальное старение [6,12,15]. Однако соответствующие связи оказались не столь прямолинейными. При БА происходит уменьшение числа лимфоцитов в периферической крови (лимфопения) по сравнению со здоровыми лицами соответствующего возраста на более поздних стадиях развития заболевания, когда формируются средний и тяжелый этапы деменции (1,2хЮ 6 и 1,1хЮ 6 , р
Оказалось также, что уровень пролиферативной активности Т-лимфоцитов при стимуляции митогенами ФГА и Кон-А у больных БА выше (р
Важным для выяснения патогенеза болезни является факт повышения продукции ИЛ-1 у больных БА, особенно если учитывать, что наиболее выраженное повышение этого показателя отличает ранние этапы болезни (10,4±2,0 и 10,9±1,2, р
Таким образом, при БА многие показатели функциональной активности иммунной системы значительно отличаются от таковых у здоровых лиц пожилого возраста. При этом выраженность изменений иммунной системы коррелирует с глубиной деменции и стадией болезни: на начальном этапе мягкой деменции соотношение лимфоцитов и их функциональная активность не изменены; у больных с умеренно выраженной деменцией на фоне лимфопении и умеренного повышения пролиферативной активности Т-лимфоцитов выявляется повышение продукции ИЛ-1; при тяжелой деменции значительная лимфопения сопровождается более резким повышением пролиферативной активности Т-лимфоцитов.
В процессе лечения пациентов с БА амиридином происходит снижение уровня пролиферативной активности лимфоцитов, независимо от направленности изменений их клинического состояния (улучшение или ухудшение), хотя улучшение психического состояния больных (особенно когнитивного статуса) сопровождалось снижением продукции ИЛ-1 после проведенной терапии, а ухудшение — ее повышением. Наибольший эффект терапии амиридином наблюдается у тех больных, уровень продукции ИЛ-1 у которых перед началом лечения был достоверно выше такового в контрольной группе здоровых. В то же время у больных БА, лечение которых не привело к улучшению клинического состояния, Уровень продукции ИЛ-1 перед началом лечения не отличался от такового в контрольной группе здоровых. На основании этих данных было сделано заключение, что показатель продукции ИЛ-1 лимфоцитами крови можно рекомендовать в качестве предиктора эффективности амиридина.
На основании изложенного можно предположить следующую последовательность «событий», происходящих в иммунной системе при БА: на первых этапах болезни, когда деменция оценивается как мягкая или средняя, в иммунной системе больного происходит повышение продукции ИЛ-1. Затем по мере углубления деменции до степени тяжелой, продукция ИЛ-1 нормализуется до возрастного уровня, но возникает активация пролиферативной активности Т-лимфоцитов, по-видимому, обусловленная накоплением ИЛ-1 в организме на предшествующих этапах болезни (ИЛ-1 активирует лимфоциты, продуцирующие ИЛ-2, который вызывает дальнейшую пролиферацию клеток).
Ключевым звеном в рассмотренном процессе, по-видимому, является повышение продукции ИЛ-1 на ранних стадиях развития болезни. Именно это изменение в функции иммунной системы отличает БА от старения в норме.
По-видимому, изменения уровня продукции ИЛ-1 может иметь отношение и к изменению протеолитической активности предшественника амилоидного белка, поскольку известно [10,14,24], что повышение синтеза ИЛ-1 в микроглиальных элементах мозга приводит к накоплению интерлейкина-6 (ИЛ-6), который стимулирует высвобождение ингибиторов этих протеаз. Этот механизм является одним из наиболее значимых патогенетических особенностей БА. В связи с этим столь большое значение приобретает дальнейшее уточнение механизмов изменений функций иммунной системы при БА, особенно активации синтеза ИЛ-1 на ранних, а возможно и доклинических, стадиях развития заболевания.
1. БутенкоГ. М., ТерешинаО. П.// Иммунология. -1992. -№ 3. — С. 15-17.
2. БутенкоГ. М.// Там же. -1993. -№ 4. — С. 4-6.
3. Девойно Л. В., ИльюченокР. Ю.//Моноаминергические системы в контроле иммунного ответа (серотонин, допамин). — Новосибирск, 1983.
4. Девойно Л. В., ИдоваГ. В., Чейдо М. А.//Бюл. экспер. биол. -1993. -№ 11. — С. 532-534.
5. Моисеева Н. Б., Чередеев А. Н.//Иммунодефициты и аллергия. — М., 1986. — С. 62-62.
6. ХаитовР. М., Вербицкий М. Ш.//Итоги науки и техники. Иммунология. — М., 1986. — Т. 14. — С. 1-165.
7. Чекнев С. Б., Ковальчук Л. В.//Геронтология и гериартрия: Иммунитет и старение. — Киев. 1987. — С. 79-83.
8. ЧекневС. Б., СаидовМ. 3., ЦветковВ. В. идр.//Иммунология. -1991. -№ 1. — С. 39-43.
9. Чередеев А. Н., Ковальчук Л. В.//Там же. -№ 2. — С. 6-14.
10. BauerJ., StraussS., Schreiter-GasserU. etal.//FEBS-1991. — Vol.285,№ l. — P. 111-114.
11. Cacabelos R., Alvarez X. F., Fernandes-Novoa L. et al.//Meth. Find. Exp. Clin. Pharmacol. -1994. — Vol. 16, -№ 2. — P. 141-151.
12. Canonica G. W., Ciprandi G., Caria A. et al.//Mech. Aging Develop. -1985. — V ol. 32, -№ 3. — P. 205-212.
13. ChopraR. K., Powers D. C., KendigN. E. et al.//Clin. Immunol. Immunopathol. -1989. — Vol. 53. — P. 297-308.
14. Del-Bo R., AngerettiN., LuccaE. etal.//Neurosci. Lett. -1995. — Vol. 188, -№ 1. — P. 70-74.
15. Doria J.//Ital. J. Med. -1988. — Vol. 4, -№ 2. — P. 83-85.
16. Ennist D. L.//Rev. Biol. Res. Aging. -1990. — Vol. 4. — P. 105-120.
17. Fabris N.// Neuroscience. -1990. — Vol. 51, -№ 4. — P. 373 — 375.
18. Griffin W. S., Sheng J. G., Roberts G. W. etal.//J. Neurophathol. E xp. Neurol. -1995. — Vol. 54, — № 2. — P. 276-281.
19. KalariaR. N., Harshbarger-Kelly. M., CohenD. L.//Neurobiol. Aging. -1996 — Vol 17 -№ 5. — P. 687-693.
20. LeonardiA., ArataL., BinoG. etal.//J. Neuroimmunol. -1989. — Vol. 22. — P. 19-22.
21. Lorens S. A., Hata N., Honda R. et al.//Neurobiol. Aging. -1990. — Vol. 11 -№ 2 — P. 139- 150.
22. McRae-Degueurce A., Booj S., Hadlid K. Et al.//Proc. Natl. Acad. Sci. USA. -1987 — Vol. 84. — P 9214-9218.
23. Singh V. K.//Mol. Chem. Neuropathol. -1996. — Vol. 28, -№ 1-3. — P. 105-111.
24. SisodiaS. S., Price D. L.//Neuroscience. -1993. — Vol. l. — P. 176-183.
25. SongL., NagelJ. E., ChrestF. J. etal.//J. CellBiochem. -1991. — Vol. 15-B. — P. 166-168.
26. SongL., Ho Kirn Y., ChopraR. K. et al.//Exp. Geronto. -1993. — Vol. 28. — P. 313-321.
27. Venero J. L., Machado A., Cano J.//Mech. Aging Develop. -1989. — Vol. 311. — P. 227-233.
источник
1.4. Болезнь Альцгеймера. Разновидности заболевания и их основные проявления. Связь болезни Альцгеймера с холинергической медиаторной системой.
Болезнь Альцгеймера относится к неизлечимым нейропсихическим заболеваниям, при которых наряду с нарушением поведенческих реакций наблюдается расстройство функций сознания. Встречается оно довольно часто и является наиболее известной причиной деменции — прогрессирующего угасания интеллектуальных функций, которая приводит к потере способности себя обслуживать. В большинстве случаев болезнь Альцгеймера не имеет генетической зависимости. Обычно она развивается в возрасте старше 65 лет, хотя может встречаться и раньше, и длится от 2 до 20 лет. Первым симптомом является потеря памяти. Заболевание неумолимо прогрессирует и заканчивается полнейшей беспомощностью.
Основным патоморфологическим признаком является дегенеративный процесс, характеризующийся потерей клеток в некоторых участках мозга (в частности, в корковом слое и гиппокампе). Многочисленные исследования направлены на выяснение причин болезни Альцгеймера. Внимание ученых привлек амилоидный -протеин (АП), основной компонент амилоидных бляшек, чрезвычайно характерных для этого заболевания. Термин «амилоидный» относится к широкой группе внеклеточных скоплений белка, обнаруженных при многих заболеваниях. Амилоидные белки, подобно крахмалу, обычно окрашиваются йодом в синий цвет, за что и получили своё название. Согласно одной гипотезе (амилоидного каскада) накопление АП вызывает патологические изменения в мозге людей с болезнью Альцгеймера. Другие изменения, наблюдающиеся при этом заболевании, такие как нейрофибриллярные клубки и изменения сосудов, являются вторичными. АП образуется из своего предшественника, названного «предшественником амилоидного белка» (ПАБ). Ген белка-предшественника локализован в 21 хромосоме, близко к месту, изменяющемуся при синдроме Дауна (трисомия 21). Наверно, поэтому больные синдромом Дауна, которые дожили до 50 летнего возраста, часто поражаются болезнью Альцгеймера.
ПАБ — это трансмембранный белок, состоящий из 770 аминокислотных остатков. АП — пептид, состоящий из 39-42 аминокислот, который отщепляется в результате протеолиза от С-конца ПАБ. Он то и образует нерастворимое внеклеточное скопление. Расщепление ПАБ могут катализировать две различные протеазы. В результате действия одной из них, получившей название секретаза, образуется растворимый фрагмент, содержащий в своем составе только часть АП (рис.18.12). Вторая является лизосомальной протеазой, в результате её действия образуется фрагмент, содержащий полную последовательность АП. Предполагается, что именно этот фрагмент накапливается во внеклеточном пространстве и приводит к формированию гистопатологических признаков, присущих болезни Альцгеймера.
Гипотеза амилоидного каскада
ПАБ может подвергаться превращению двумя путями. Первый включает секретазу, действие которой приводит к образованию пептидов, которые не содержат полную аминокислотную последовательность амилоидного -протеина (АР). Эти пептиды раствориы в воде и не осаждаются с образованием амилоида. В соответствии со вторым путем действие эндосомальных-лизосомальных протеаз(ы) приводит к образованию амилоидного-белка или пептидов, содержащих его полную аминокислотную последовательность в своем составе. Такие пептиды выпадают в осадок с образованием амилоида. Предполагается, что это приводит к формированию нейрофибриллярных клубочков и смерти клетки.
В некоторых случаях болезни Альцгеймера, особенно с ранним началом заболевания, были обнаружены мутации на С-конце ПАБ. Белковые фрагменты, которые образовываются в результате таких мутаций, содержат в своем составе АП. Однако роль этих фрагментов до настоящего времени неясна.
Второй часть гипотезы амилоидного каскада в развитии болезни Альцгеймера основывается на том, что АП и фрагменты, содержащие этот белок, прямо или косвенно являются нейротоксичными веществами. Замечено, что в нейронах в присутствии АП может увеличиваться внутриклеточная концентрация Са2+. Уровень же Са2+ способен оказывать регуляторное влияние на активность некоторых протеинкиназ, которые катализируют фосфорилирование tau-белка. Таким образом, повышение уровня Са2+ может привести к гиперфосфорилированию tau и образованию спаренных спирализованных нитей, характерных для нейрофибриллярных клубочков.
Гипотетическая схема последовательности событий, вовлекаемых в развитие некоторых случаев болезни Альцгеймера
Мутации гена ПАБ обнаружены только в небольшом количестве случаев этого заболевания, поэтому равновероятно существование и других патогенетических механизмов.
Гипотеза зачастую хороша не тем, что она правильная, а тем, что она стимулирует дальнейшие исследования. В частности, ключевым моментом, требующим в дальнейшем выяснения, является ответ на вопрос, первично ли накопление АП в развитии болезни Альцгеймера или это вторичное событие, которое развивается вследствие неизвестных ещё более ранних явлений? Существуют и другие гипотезы происхождения этого заболевания, которые представляются менее вероятными на сегодняшний день. Так, было замечено, что в составе бляшек при болезни Альцгеймера часто содержится повышенное количество алюминия. Это позволило предположить, что одной из причин заболевания является увеличенное поступление его в организм с пищей. Одновременно возник вопрос, в действительности ли увеличенное отложение алюминия в бляшках вызывает болезнь Альцгеймера или уже поврежденные каким-то другим процессом клетки приобретают способность усиленно поглощать алюминий? Ответа на этот вопрос на сегодняшний день нет. С другой стороны, в ткани мозга людей, умерших от болезни Альцгеймера, часто обнаруживается существенное снижение ацетилхолина и других нейромедиаторов, однако эти изменения представляются вторичными, возникшими вследствие повреждения клеток при этом заболевании.
С раскрытием механизмов развития болезни Альцгеймера связывают разработку эффективных тестов его диагностики и лечения. К примеру, большое значение могли бы иметь соединения, которые бы ингибировали образование АП или делали бы его растворимым в воде. В настоящее время точный дигноз болезни Альцгеймера чаще ставится на аутопсии при обнаружении характерных бляшек. Перспективной представляется разработка теста, основанного на определении количества ПАБ в цереброспинальной жидкости (ЦСЖ) с помощью специфических моноклональных антител. Считают, что в ЦСЖ людей, страдающих этим заболеванием, его уровень существенно (
в 3 раза) ниже, чем у здоровых людей. В настоящее время нет специфической лекарственной терапии болезни Альцгеймера, хотя все более и более реальной становится возможность с помощью препаратов влиять на накопление АП в тканях. Такой перспективой, в частности, обладает выделенный из мозга фактор роста нервов. В некоторых областях мозга больных его количество снижено. Сейчас изучается терапевтический эффект от его использования у экспериментальных животных, у которых хирургическим путем была вызвана нейрональная дегенерация.
1.5. Шизофрения. Наследственная предрасположенность, основные проявления и биохимические основы шизофрении. Рассогласованность в работе основных нейромедиаторных систем как основа психических нарушений при шизофрении.
Шизофренические расстройства относятся к умственным расстройствам. Для них характерна психотическая симптоматика, отражающая изменения со стороны мышления, чувств и поведения. Причина или причины шизофрении на сегодняшний день неизвестны.
Проведенные генетические исследования показали большое значение генетического фактора в происхождении шизофрении. К примеру, вероятность развития шизофрении у ребенка, у которого оба родителя имеют это заболевание, составляет 39%. Среди монозиготных близнецов заболеваемость шизофренией совпадает в 47% случаев. Однако неизвестно, является ли шизофрения моногенным, полигенным или многофакторным состоянием.
Поиск генов, ответственных за развитие этого заболевания, привел к противоречивым результатам. Сначала было обнаружено, что соответствующий локус находится на 5 хромосоме. Однако эти данные не нашли подтверждения в последующих исследованиях, и вопрос остается открытым.
Структурные изменения в мозге больных шизофренией
В мозге многих больных шизофренией обнаруживаются изменения в медиальной лобной доле (парагиппокампальная извилина, гиппокамп и миндалина). Эти зоны участвуют в сборе и обработке информации из коры.
Дофаминовая гипотеза происхождения шизофрении
В различные периоды времени возникали биохимические теории, в соответствии с которыми в возникновении шизофрении участвовали ацетилхолин, -аминомасляная кислота (ГАМК), норадреналин, опиаты, пептиды и другие молекулы. Однако в последние 30 лет наибольшее внимание приковано к дофамину. В начале 50-х годов, сразу после успешного начала использования неролептиков (антипсихотиков) для лечения психозов, в том числе шизофрении, было замечено, что у шизофреников в ходе такой терапии развивается паркинсонизм. Подобные наблюдения навели на мысль о том, что нейролептики снижают уровень дофамина в организме. Эти и другие факты подтверждали участие дофамина в развитии шизофрении (табл. 18.10). В соответствии с гипотезой происхождения шизофрении эту патологию рассматривают как проявление гипердофаминергии. Противоположно, болезнь Паркинсона может рассматриваться как состояние гиподофаминергии.
Аргументы в пользу дофаминергической гипотезы происхождения шизофрении
Нейролептики (антипсихотики) часто вызывают паркинсонизм, что привело к заключению об их способности снижать уровень дофамина.
Действие нейролептиков направлено на снижение биологической активности дофамина в мезолимбических дофаминовых нейронах.
Другие лекарственные препараты (в частности, L-ДОФА, амфетамин), которые оказывают дофамин-миметическое действие на его метаболизм, вызывают симптомы шизофрении.
Длительное лечение нейролептиками приводит к снижению уровня гомованилиновой кислоты в цереброспинальной жидкости и улучшению клинического состояния больных.
Выраженность антипсихотического действия большинства нейролептиков коррелирует с их связыванием с D2 рецепторами.
При анализе трупного материала и результатов позитронной томографии отмечено, что в мозге больных шизофренией увеличена плотность D2 рецепторов.
Результаты биохимических исследований относительно выяснения роли дофамина в патогенезе шизофрении можно разделить на три группы.
1) Определение количества дофамина в ткани мозга. Большинство исследователей обнаружили его увеличение, хотя полученные ими данные значительно варьировали.
2) Определение метаболитов дофамина в ткани мозга и биологических жидкостях. Особое внимание было уделено гомованилиновой кислоте как наиболее важному метаболиту у человека. У больных шизофренией её уровень оказался существенно повышенным, он снижался в ходе лекарственной терапии. Однако данные опять значительно варьировали.
3) Определение дофаминовых (D) рецепторов. Количество D2 рецепторов увеличено в мозге при шизофрении. Важным обстоятельством явилось установление зависимости между выраженностью антипсихотического действия нейролептиков и их способностью конкурировать in vitro с дофамином за связывание с D2 рецепторами.
Получив такие данные, неудивительно, что усилия исследователей сосредоточились на дофаминовых рецепторах. Благодаря технологии клонирования генов, удалось обнаружить их 5 различных классов. Все они являются трансмембранными белками — гликопротеинами, сопряженными с G-белками. Рецепторы D2, D3 и D4 очень похожи между собой.
Все эти данные позволили несколько изменить первоначальную гипотезу. В настоящее время полагают, что для шизофрении характерно нарушение дофаминергической активности, которое не всегда сводится к её увеличению. В некоторых областях мозга дофаминергическая активность действительно может быть повышена, тогда как в других она в это же время снижена. Следует иметь в виду, что в развитии шизофрении могут принимать участие и другие нейромедиаторы, например, серотонин, самостоятельно или путем взаимодействия с дофаминергическими системами.
источник
Определение. Болезнь Альцгеймера (син.: деменция альцгеймеровского типа) — это хроническое прогрессирующее дегенеративное заболевание головного мозга, которое проявляется нарушениями памяти и других когнитивных функций.
Болезнь Альцгеймера (БА) названа в честь немецкого психиатра и нейроморфолога, который в 1907 г. описал случай деменции у 56-летней женщины. За 5 лет до смерти у нее появились симптомы прогрессирующей потери памяти, она начала путаться в окрестностях, а потом и в собственном доме. У нее также отмечались бред преследования и расстройства речи, чтения и письма. Патоморфологическое исследование выявило атрофию головного мозга, особые нейрональные изменения (нейрофибриллярные сплетения) и множественные милиарные очаги (сенильные или невритические бляшки). А. Альцгеймер особо подчеркнул пресенильный характер заболевания, считая, что речь идет о новом заболевании, отличном от сенильной деменции [11]. Его руководитель Э.Крепелин поддержал эту точку зрения и в очередном издании своего руководства по психиатрии, выпущенном в 1910 г., предложил называть данный пресенильный тип деменции болезнью Альцгеймера, выделив его в самостоятельную нозологическую единицу [20]. Однако в 1911 г. сам А. Альцгеймер высказал суждение, что описанное им заболевание является атипичной формой сенильной деменции.
В то время нейродегенеративный процесс считался относительно редкой причиной деменции. Полагали, что главной причиной слабоумия в пожилом и старческом возрасте является поражение сосудов головного мозга — так называемая атеросклеротическая деменция. Однако выявление на аутопсии характерных для БА патоморфологических изменений у большинства больных с деменцией привело к активному изучению данного заболевания во второй половине XX века. При этом патоморфологические изменения при сенильной и пресенильной формах первичной дегенеративной деменции оказались одинаковыми. Поэтому, исходя из единства клиники и морфологии, пресенильная и сенильная деменции были объединены в единую нозологическую форму под эпонимическим обозначением болезнь Альцгеймера [1, 3, 4, 17, 29].
Эпидемиология и факторы риска. Болезнь Альцгеймера является одним из наиболее распространенных нейродегенеративных заболеваний и самой частой причиной деменции в популяции. Данное заболевание вызывает не менее 35–40 % деменций. Распространенность БА в возрастном диапазоне от 65 до 85 лет составляет от 2 до 10 %, а среди лиц старше 85 лет — 25 %. В настоящее время в мире проживает около 24 млн. пациентов с БА [1, 3, 4, 16, 17, 22, 24].
Пожилой возраст является наиболее сильным фактором риска БА. Пик заболеваемости БА приходится на 80–90 годы жизни: переход через 80-летний рубеж утраивает риск развития данного заболевания [1, 3, 4, 16, 17, 22, 29].
Большое значение имеет также семейный анамнез по данному заболеванию, особенно при его начале в возрасте до 65 лет. Считается, что риск развития БА в 4 раза выше у близких родственников больных и в 40 раз — при наличии в роду двух и более случаев деменции. По эпидемиологическим данным, около 30 % больных с БА имеют родственников, болевших БА. Наличие в семейном анамнезе указаний на возникновение синдрома Дауна также является фактором риска развития БА [3, 7, 15, 16, 24].
К другим факторам, повышающим риск развития БА, относятся [17, 24, 19, 29]:
• неконтролируемая артериальная гипертензия в среднем и пожилом возрасте;
• атеросклероз магистральных артерий головы;
• хроническая гипоксия, например, при заболеваниях дыхательных путей;
• черепно-мозговая травма в анамнезе;
• низкий уровень образования;
• низкая интеллектуальная активность в течение жизни;
• эпизоды депрессии в молодом и среднем возрасте;
Этиология. БА является заболеванием с многофакторной этиологией. Согласно современным представлениям, существует генетическая предрасположенность к БА, однако для ее клинической реализации необходимо также неблагоприятное воздействие внешних средовых факторов.
Семейные формы заболевания встречаются относительно нечасто — не более 10 % всех случаев. Семейные формы БА, как правило, характеризуются ранним началом (до 65 лет), аутосомно-доминантным типом наследования и высокой пенетрантностью патологического гена. Недавние исследования в области генетики позволили идентифицировать три гена, ответственных за развитие семейных форм БА с ранним началом [7, 15, 17]:
• ген, кодирующий предшественник амилоидного белка (21 — я хромосома);
• пресенилин-1 (14-я хромосома);
Наиболее частой является мутация гена пресенилин-1 на 14-й хромосоме, которая встречается в 60–70 % всех пресенильных случаев семейной БА. Наличие данной мутации означает почти 100 % вероятность заболеть БА в возрастном диапазоне от 40 до 65 лет [7, 15, 17].
Мутация гена, кодирующего предшественник амилоидного белка (ПАБ) на 21-й хромосоме, встречается у 3–5% всех семей с пресенильным типом заболевания. С этим геном связана, в частности, БА при болезни Дауна. Известно, что у пациентов с болезнью Дауна в возрасте 20–30 лет на фоне изначальной умственной отсталости развивается деменция, патоморфология которой удовлетворяет диагностическим критериям БА [7, 15, 17].
Наиболее редкой является мутация в гене пресенилин-2 на 1-й хромосоме, которая характеризуется низкой пенетрантностью (низкая заболеваемость, несмотря на наличие патологического гена). Мутации гена пресенилин-2 встречаются не только при раннем начале БА, но и при сенильных формах данного заболевания [7, 15, 17].
Ген ПАБ, пресенилин-1 и пресенилин-2 кодируют белки, участвующие в метаболизме предшественника амилоидного белка (см. раздел «Патогенез»).
В возникновении поздней семейной и спорадической формы БА решающее значение придается наличию аллеля аполипопротеина Е4 (апоЕ4) на 19-й хромосоме. АпоЕ является белком, участвующим в транспорте липидов, и имеет большое значение в биосинтезе клеточных мембран. В человеческом геноме могут присутствовать 4 аллеля гена апоЕ. Наличие гена апоЕ4 увеличивает риск развития БА примерно в 2 раза по сравнению со среднестатистическим риском. Присутствие гена апоЕ2, напротив, уменьшает риск возникновения БА [7, 15, 17].
Активно изучается этиологическая роль других генетических факторов, так как БА может развиваться в отсутствие четырех приведенных выше известных патологических генов.
Как уже указывалось выше, только носительства патологических генов в большинстве случаев недостаточно для прижизненной реализации врожденной генетической программы. Первостепенно важную роль играет пожилой возраст. Увеличивают темп дегенеративного процесса и приближают время клинической манифестации симптомов деменции церебральная ишемия и гипоксия, черепно-мозговая травма и дисметаболические нарушения (в том числе дефицит витаминов группы В, фолиевой кислоты, гипотиреоз, печеночная и почечная недостаточность и др.).
Патогенез. Согласно наиболее обсуждаемой на сегодняшний день «амилоидной гипотезе», отправной точкой патогенеза БА является нарушение метаболизма ПАБ. Установленные в настоящее время гены БА либо непосредственно кодируют данный белок (ген, кодирующий ПАБ, 21-я хромосома), либо метаболизирующие его ферменты (так называемые альфа-, бета- и гамма-секретазы: пресенилин-1, хромосома 14 и пресенилин-2, хромосома 1).
В норме ПАБ расщепляется ферментом альфа-секретазой на одинаковые по величине полипептиды, которые не являются патогенными. При генетической дефектности данного белка иди дефектности ферментных систем, ПАБ расщепляется бета- и гамма-секретазами на различные по длине фрагменты. При этом длинные фрагменты (альфа-бета-42) являются нерастворимыми и поэтому откладываются в паренхиме головного мозга и стенках церебральных сосудов (стадия диффузного церебрального амилоидоза). Далее в паренхиме головного мозга происходит агрегация нерастворимых фрагментов в патологический белок — бета-амилоид. «Гнездные» отложения данного белка в паренхиме головного мозга называют сенильными бляшками. Отложение амилоидного белка в церебральных сосудах приводит к развитию церебральной амилоидной ангиопатии, которая является одной из причин хронической ишемии мозга [1, 3,10, 17, 22, 26, 29].
Бета-амилоид и нерастворимые фракции диффузного амилоидного белка обладают нейротоксическими свойствами. В эксперименте показано, что на фоне церебрального амилоидоза активируются тканевые медиаторы воспаления, усиливается выброс возбуждающих медиаторов (глутамат, аспартат и др.), повышается образование свободных радикалов. Результатом всего этого сложного каскада событий является повреждение нейрональных мембран, индикатором которого является образование внутри клеток нейрофибриллярных сплетений (НФС). НФС представляют собой фрагменты биохимически измененной внутренней мембраны нейрона и содержат гиперфосфорилированный тау-протеин. В норме тау-протеин является одним из основных белков внутренней мембраны нейронов. Наличие внутриклеточных НФС свидетельствует о необратимом повреждении клетки и ее скорой гибели, после которой НФС выходят в межклеточное пространство («НФС-призраки»). В первую очередь и в наибольшей степени страдают нейроны, окружающие сенильные бляшки [1, 3, 10, 17,12, 26, 29] (рис. 3.1).
Следует отметить, что начальные признаки альцгеймеровской дегенерации, такие как диффузный церебральный амилоидоз и даже сенильные бляшки, обнаруживаются у подавляющего большинства старых людей с нормальными по возрасту когнитивными функциями. Поэтому обязательным морфологическим критерием диагноза БА является присутствие не только ранних, но и поздних признаков БА, таких как НФС и гибель нейронов. Имеет значение также выраженность изменений. При этом степень когнитивных нарушений коррелирует с уменьшением числа нейронов и синапсов между ними и не коррелирует с выраженностью церебрального амилоидоза [12, 17, 26, 29].
Наличие другого сопутствующего патологического процесса в головном мозге, даже незначительно выраженного, ведет к клинической манифестации синдрома деменции на более ранних этапах дегенеративного процесса. Речь идет в первую очередь о сосудистой мозговой недостаточности, которая укорачивает доклиническую фазу БА и переводит бессимптомный процесс в симптомный. Вероятно, поэтому БА разделяет с цереброваскулярной патологией общие факторы риска (артериальная гипертензия, атеросклероз, гиперлипидемия, гипергомоцистеинемия, сахарный диабет), а своевременная коррекция указанных нарушений ведет к отсрочке наступления деменции [3,19, 22].
Патологическая анатомия и нейрохимия БА. Патологическая анатомия БА представлена тремя основными видами изменений: сенильными бляшками, внутриклеточными нейрофибриллярными сплетениями и церебральной атрофией. Церебральная атрофия проявляется уменьшением объема и массы мозга, расширением корковых борозд и желудочковой системы [2, 12, 18, 26].
Различные отделы головного мозг вовлекаются в патологический процесс при БА неодинаково. Наибольшая выраженность атрофических изменений отмечается в гиппокампе и функционально связанных с ним глубинных отделах височных долей головного мозга. Затем патологические изменения последовательно развиваются в задних отделах височных долей и в теменных долях головного мозга. Наиболее поздно в патологический процесс вовлекаются конвекситальные отделы лобной коры и первичные моторные и сенсорные зоны [4, 12, 18, 26].
Важную роль в формировании когнитивных и поведенческих симптомов БА играют изменения со стороны нейротрансмиттерных систем. Относительно ранним событием патогенеза БА является поражение ядра Мейнерта и безымянного вещества. Данные образования являются началом восходящих ацетилхолинергических путей в различные отделы головного мозга. Гибель пресинаптических ацетилхолинергических нейронов данных анатомических структур приводит к недостаточности ацетилхолинергической нейротрансмиттерной системы. Имеется прямое соответствие между тяжестью деменции и центральным ацетилхолинергическим дефицитом. Снижение содержания ацетилхолина и уменьшение плотности рецепторов к ацетилхолину выявляются в гиппокампе, височной, теменной, лобной и орбитофронтальной коре. Помимо ацетилхолинергической системы, при БА страдают также другие нейротрансмиттерные системы: глутаматергическая, норадренергическая, дофаминергическая, серотонинергическая и др. [3, 4, 13, 24].
Клиническая картина. Дегенеративный процесс при БА начинается приблизительно за 15–20 лет до появления клинических симптомов. Первым и ведущим проявлением заболевания чаще всего являются нарушения памяти. Нередко они возникают и текут изолированно от других когнитивных нарушений за несколько лет до развития деменции. В первую очередь нарушается память на текущие или недавно произошедшие события, в то время как воспоминания о давних событиях остаются временно сохранными. Такая закономерность прогрессирования нарушений памяти при БА получила название закона Рибо. Пациенты не удерживают в памяти текущую информацию, что вызывает затруднения в повседневной деятельности. Это обусловлено дефектом кодирования информации, перевода ее в долговременную память и недостаточностью извлечения информации. Нарушения памяти довольно рано приводят к нарушению ориентировки во времени (амнестическая дезориентировка во времени). С развитием заболевания нарушается память и на отдаленные события. Иногда «пустоты» в памяти заменяются вымышленными событиями (так называемые конфабуляции — ложные воспоминания). На продвинутых стадиях заболевания пациенты могут припомнить лишь самые важные события жизни [5, 14, 25].
Нарушения памяти по преимуществу носят модально-неспецифический характер, хотя в большей степени и раньше страдает зрительная память и память на запахи, что соответствует локализации и динамике патоморфологических изменений в головном мозге [5, 14, 25].
Для исследования мнестической функции используются нейропсихологические тесты с запоминанием и воспроизведением серии слов и изображений. БА характеризуется особым видом нарушений памяти, отражающим заинтересованность гиппокампа и глубинных отделов височных долей головного мозга. При этом наблюдаются значительная разница между непосредственным и отсроченным от предъявления воспроизведением (повышенная чувствительность следа памяти к интерференции), посторонние вплетения (нарушение избирательности памяти), неэффективность организации материала на этапе заучивания и подсказок при воспроизведении [5, 8, 14].
Нарушения речи довольно часты у больных БА и могут наблюдаться на относительно ранних этапах течения болезни. Они частично связаны с базисными нарушениями памяти и проявляются затруднениями в назывании предметов и объектов (аномия, амнестическая афазия) и восприятии чужой и собственной речи (сенсорная, семантическая афазия). Это проявляется в пробе на называние слов одной семантической категории в ограниченный интервал времени (например, назвать животных, растения и т. д.). Затруднения в подборе слов могут маскироваться их заменой близкими по смыслу, иногда возникают парафазии (произнесение слов, не соответствующих контексту высказывания). Письмо, чтение и повторение слов поначалу остаются сохранными. По мере развития заболевания затруднения в назывании и подборе слов нарастают, учащаются парафазии, нарушаются понимание речи, письмо, чтение, речь больного утрачивает смысл. Нередко появляются эхолалии (повторение чужих слов) или палилалии (повторение собственных слов). В конце концов происходит полное нарушение речевых функций с непониманием обращенной речи и отсутствием спонтанной речи — развивается тотальная афазия. Артикуляция сохраняется до последних этапов заболевания, в финале оказываются возможными нечленораздельные высказывания или развивается мутизм [8, 14, 25, 28, 29].
Зрительно-пространственные дисгностические и диспрактические нарушения являются обязательными, часто рано развивающимися, и могут быть ведущими проявлениями БА. Поначалу наблюдаются затруднения в ориентировке в незнакомой местности или обстановке, продумывании схемы поездок на транспорте, особенно в метро, когда требуется пользоваться схемами. Позднее развивается выраженная дезориентировка в пространстве, даже в относительно знакомых местах [8, 14, 25, 28, 29].
В клинической практике для тестирования пространственных функций пациента просят перерисовывать сложные геометрические фигуры или нарисовать циферблат часов со стрелками. Трудности при выполнении этих заданий, которые свидетельствуют о пространственных расстройствах, называются пространственной (конструктивной) апраксией. Пространственная апраксия почти всегда сочетается с пространственной агнозией, так как в их основе лежат общий механизм (утрата представлений о трехмерном пространстве) и общий субстрат (патология теменных долей головного мозга). Поэтому иногда пространственные расстройства объединяют термином «апракто-агностический синдром». На поздних этапах болезни прогрессирование диспрактических нарушений ведет к нарушениям самообслуживания, в частности нарушениям одевания (апраксия одевания) [14, 25, 28, 29].
На ранних этапах заболевания критика к своему состоянию полностью или частично сохранна. Осознание прогрессирующего когнитивного дефекта часто вызывает обоснованную тревогу и беспокойство. В большинстве случаев пациенты выглядят растерянными, активно жалуются на снижение Памяти, могут предъявлять другие жалобы, отражающие повышенный Уровень тревоги. В 25–40 % случаев развивается депрессия, в структуре которой почти всегда присутствуют выраженные тревожные нарушения [22, 24].
По мере прогрессирования заболевания снижается критика и происходит так называемая сенильная перестройка структуры личности. У пациентов появляются эгоцентризм, ворчливость, склонность к подозрениям и конфликтам. Позднее на фоне личностных изменений наблюдается склонность к бредообразованию. Весьма специфичным для развернутых стадий БА видом поведенческих нарушений является бред ущерба: пациент подозревает ближайших родственников в том, что они крадут его вещи, собираются оставить без помощи, пытаются уморить и т. д. Регулярно встречаются и другие виды поведенческих нарушений: бесцельная двигательная активность (бродяжничество, хождения из угла в угол, перекладывание вещей), изменение пищевого поведения (гиперфагия, повышенная тяга к сладкому), сексуальная несдержанность. На этапе выраженной деменции возникает так называемый симптом зеркала: больные перестают узнавать свое изображение в зеркале, воспринимают его как постороннего человека. Бред и другие виды поздних поведенческих нарушений не являются обязательными для БА, но развиваются у большинства пациентов с этим заболеванием [8, 22, 24].
Прогрессирование когнитивных и поведенческих нарушений закономерно приводит к трудностям в повседневной жизни и к постепенной утрате независимости и самостоятельности. На начальных этапах БА нарушаются наиболее сложные виды повседневной деятельности, такие как работа, хобби и увлечения, социальная активность, общение с другими людьми. При этом у себя дома пациент полностью адаптирован, может ходить в ближайший магазин и совершать путешествия по хорошо знакомым маршрутам. Позднее возникают трудности у себя дома, развивается частичная, а затем и полная зависимость от посторонней помощи. На стадии тяжелой деменции бред и другие поведенческие расстройства постепенно регрессируют из-за грубой интеллектуальной недостаточности. Больные апатичны и не предпринимают каких-либо попыток активной деятельности. Снижаются чувства голода и жажды. В финале БА речь утрачивается, пациенты не могут ходить и поддерживать равновесие, испытывают трудности при кормлении из-за нарушения жевания. Смерть наступает из-за осложнений обездвиженности или от сопутствующих заболеваний [3, 4, 8, 21, 24].
Описание основных клинических характеристик БА приведено в «Общей шкале нарушений» (Global deterioration rating, Reisberg В., 1982) (Приложение 11) [27].
Вплоть до наиболее поздних стадий в подавляющем большинстве случаев БА отсутствуют двигательные, чувствительные и тазовые нарушения. Редко (не более чем в 10 % случаев) выявляются легкие экстрапирамидные симптомы: гипокинезия и повышение мышечного тонуса. БА с экстрапирамидными симптомами иногда выделяют в особую форму заболевания, которая характеризуется более быстрым темпом прогрессирования. Предполагается, что морфологической основой БА с экстрапирамидными симптомами является сочетание нейродегенеративных изменений, характерных для БА (сенильные бляшки, НФС), с тельцами Леви, которые являются морфологическими признаками болезни Паркинсона или деменции с тельцами Леви [3, 4, 22, 23].
На стадии тяжелой деменции в неврологическом статусе определяются нарушения походки, связанные с утратой навыка ходьбы (апраксия ходьбы). Утрачивается также контроль над мочеиспусканием и дефекацией. У части пациентов развиваются миоклонии [3, 4, 21, 22]. Особенности клинической картины БА приведены в таблице 3.1.
Основные клинические характеристики БА
| Дебют БА (легкая деменция) | Развернутые стадии (умеренная деменция) | Поздние стадии (тяжелая деменция) | |
|---|---|---|---|
| Когнитивные расстройства | Нарушения памяти на недавние события. Отдаленная память сохранена. Амнестическая дезориентировка во времени. Нарушения ориентировки в незнакомой местности. Трудности называния предметов | Выраженные нарушения памяти: вспоминает лишь главные события жизни. Дезориентировка в месте и времени. Апракто-агностический синдром. Амнестическая, позже сенсорная афазия | Отсутствие когнитивной деятельности, утрата речи |
| Эмоциональные и поведенческие расстройства | Тревожно-депрессивные расстройства | Подозрительность, бред ущерба, агрессивность, галлюцинации | Апатия, снижение витальных мотиваций |
| Неврологический статус | Нет нарушений | Нет нарушений. Редко: гипокинезия, повышение тонуса по пластическому типу | Нарушения походки и мочеиспускания. Редко: миоклонии |
| МРТ головы | Атрофия гиппокампа | Диффузная атрофия с акцентом на теменно-височные отделы | Грубая диффузная церебральная атрофия |
Согласно МКБ-10 выделяют пресенильную и сенильную формы БА. О пресенильной форме БА говорят при начале заболевания в возрасте 65 лет, а о сенильной форме — при начале после 65 лет. Пресенильная БА характеризуется более быстрым прогрессированием и ранним присоединением афазии, апраксии и агнозии, в большинстве случаев можно проследить семейный анамнез заболевания. Сенильная БА прогрессирует медленнее, нарушения памяти длительное время остаются ведущим симптомом, в то время как другие когнитивные нарушения представлены мягко, семейный анамнез обычно не прослеживается (см. табл. 3.2) [6].
Диагноз. Диагностика БА базируется на характерных анамнестических, клинических и инструментальных данных. Прижизненный диагноз всегда носит вероятностный характер: достоверный диагноз БА может быть установлен только на основании патоморфологического исследования.
Анамнестически БА характеризуется незаметным началом: пациент и его родственники с трудом определяют время появления первых симптомов. Заболевание носит неуклонно прогрессирующий характер. Наибольший темп прогрессирования отмечается на стадиях легкой и умеренной деменции. На стадии тяжелой деменции темп прогрессирования уменьшается, иногда симптомы носят почти стационарный характер. Следует отметить, что, хотя длительные остановки прогрессирования заболевания считаются нехарактерными для БА, они все же не исключают данный диагноз, особенно у лиц пожилого и старческого возраста [5, 23, 28].
Различия между пресенильной и сенильной БА
| Пресенильная БА | Сенильная БА | |
|---|---|---|
| Семейный анамнез | Часто | Редко |
| Клиника | Нарушения памяти + выраженные афазия, апраксия, агнозия | Доминируют нарушения памяти |
| Прогрессирование | Быстрое | Медленное, возможны периоды стабилизации (плато) |
Основным диагностическим признаком БА является характерная клиническая картина деменции: нарушения памяти преимущественно на недавние события в сочетании с другими когнитивными расстройствами в отсутствие очаговой неврологической симптоматики. Диагностические критерии БА в соответствии с Международной классификацией болезней (10-го пересмотра) предусматривают следующее [6]:
• Клинический диагноз деменции, который включает:
— нарушения памяти, которые проявляются в нарушении способности к запоминанию нового материала, а в более тяжелых случаях — также в затруднении припоминания ранее усвоенной информации. Нарушения проявляются как в вербальной, так и в невербальной модальности. Мнестические расстройства должны быть объективизированы с помощью нейропсихологических тестов;
— нарушение других когнитивных функций, что проявляется нарушением способности к суждениям, мышлению (планирование, организация) и переработке информации. Эти нарушения должны быть объективизированы, желательно с использованием соответствующих нейропсихологических тестов. Необходимым условием диагноза деменции является снижение когнитивных функций по сравнению с более высоким исходным мнестико-интеллектуальным уровнем;
— нарушение когнитивных функций определяется на фоне сохранного сознания;
— нарушение эмоционального контроля или мотиваций или изменение социального поведения — по меньшей мере одно из следующих: эмоциональная лабильность, раздражительность, апатия, асоциальное поведение;
— для достоверного диагноза перечисленные признаки должны наблюдаться по меньшей мере в течение 6 мес.; при более коротком наблюдении диагноз может быть предположительным.
• Отсутствуют анамнестические, физикальные и инструментальные данные о другом заболевании, которое может вызывать деменцию (цереброваскулярное поражение, ВИЧ, болезнь Паркинсона, хорея Гентингтона, нормотензивная гидроцефалия, системные заболевания, гипотиреоз, дефицит витамина В12 или фолиевой кислоты, гиперкальциемия, алкоголизм, наркомания).
В клинической практике и при проведении научных исследований, наряду с диагностическими критериями МКБ-10, широко используются также диагностические критерии Американского национального института неврологических и коммуникативных расстройств и инсульта и Общества болезни Альцгеймера и ассоциированных расстройств (NINCDS-АDRA), которые приведены в таблице 3.3 [23].
Применение диагностических критериев NINCDS-АDRA позволило добиться соответствия клинической и патоморфологической диагностики БА в 85–95 % случаев. Однако нередкой остается неточная клиническая диагностика, когда другие неврологические, соматические и психиатрические заболевания, сопровождающиеся или проявляющиеся деменцией, диагностируются как БА. Как правило, диагностика заболевания опаздывает на 2–3 года после появления его первых клинических признаков.
Лабораторные и инструментальные методы исследования имеют вспомогательное значение в диагностике БА. Рутинные анализы крови, мочи и спинномозговой жидкости не выявляют какой-либо патологии. В то же время определение в спинномозговой жидкости специфических маркеров дегенеративного процесса может служить дополнительным подтверждением клинического диагноза. В качестве таких маркеров в настоящее время рассматривается содержание в спинномозговой жидкости фрагмента амилоидного белка (альфа-бета-42) и тау-протеина. БА характеризуется уменьшением концентрации альфа-бета-42 и одновременным увеличением концентрации тау-протеина. Данный признак имеет особенно важное диагностическое значение на додементных стадиях БА, когда клиническая диагностика не может быть достаточно надежной в силу мягкости симптоматики [22, 24, 26, 28, 29].
Диагностические критерии БА национального (США) Института неврологических и коммуникативных расстройств и инсульта и Общества болезни Альцгеймера и ассоциированных расстройств (NINCDS-ADRDA) (McKаhn G. et al., 1984)
| Определенная БА: |
| • клиническая картина, соответствующая «вероятной БА» (см. ниже); |
| • гистопатологические признаки БА, полученные при биопсии или при патоморфологическом исследовании. |
| Вероятная БА: |
| А. Обязательные признаки: |
| 1. Наличие деменции по результатам скрининговых нейропсихологических шкал. |
| 2. Наличие нарушений не менее чем в двух когнитивных сферах или наличие прогрессирующих нарушений в одной когнитивной сфере. |
| 3. Прогрессирующий характер нарушений памяти и других когнитивных функций. |
| 4. Отсутствие нарушений сознания. |
| 5. Начало заболевания в возрастном диапазоне от 40 до 90 лет. |
| 6. Отсутствие признаков системных дисметаболических нарушений или других заболеваний головного мозга, которые объясняли бы нарушения памяти и других когнитивных функций. |
| Б. Дополнительные диагностические признаки: |
| 1. Наличие прогрессирующей афазии, апраксии или агнозии. |
| 2. Трудности в повседневной жизни или изменение поведения. |
| 3. Наследственный анамнез БА. |
| 4. Отсутствие изменений при рутинном исследовании спинномозговой жидкости. |
| 5. Отсутствие изменений или неспецифические изменения (например, увеличение медленноволновой активности) при электроэнцефалографии. |
| 6. Признаки нарастающей церебральной атрофии при повторных КТ- или МРТ- исследованиях головы. |
| В. Признаки, не противоречащие диагнозу БА (после исключения других заболеваний ЦНС): |
| 1. Периоды стабилизации симптоматики. |
| 2. Симптомы депрессии, нарушения сна, недержание мочи, бред, галлюцинации, иллюзии, вербальное, эмоциональное или двигательное возбуждение, потеря веса. |
| 3. Неврологические нарушения (на поздних стадиях болезни) — повышение мышечного тонуса, миоклонии, нарушение походки. |
| 4. Эпилептические припадки (на поздних стадиях болезни). |
| 5. Нормальная КТ- или МРТ-картина. |
| 6. Необычное начало, клиническая картина или история развития деменции. |
| 7. Наличие системных дисметаболических расстройств или других заболеваний головного мозга, которые, однако, не объясняют основной симптоматики. |
| Г. Признаки, исключающие диагноз БА: |
| 1. Внезапное начало деменции. |
| 2. Очаговая неврологическая симптоматика (например, гемипарез, нарушение полей зрения, мозжечковая атаксия). |
| 3. Эпилептические припадки или нарушения ходьбы на ранних стадиях заболевания. |
| Возможная БА: |
| • атипичное начало, течение и симптоматика деменции при отсутствии других ее причин (неврологических, психиатрических, соматических); |
| • наличие соматических заболеваний и/или органического поражения головного мозга, которые могут вызвать деменцию, но не рассматриваются в качестве ее причины (в данном случае). |
Диагностическое значение электрофизиологических методов исследования невелико. Обычно ЭЭГ фиксирует увеличение медленноволновой активности, особенно в задних отделах коры головного мозга. Весьма характерно также удлинение латентных периодов поздних компонентов когнитивных вызванных потенциалов, отражающих процессы внимания и принятия решения (Р300). Однако указанные изменения неспецифичны и наблюдаются также при когнитивных нарушениях иной природы, расстройствах функционального ряда [3, 4, 23, 28].
Обязательным этапом обследования пациентов с БА является нейровизуализация: компьютерная рентгеновская или магнитно-резонансная томография головного мозга. Целью нейровизуализации является, во-первых, исключение других поражений головного мозга с клиникой деменции и, во- вторых, получение дополнительных позитивных подтверждений диагноза. Первая задача считается более важной: диагноз БА остается правомерным и при отсутствии каких-либо специфических нейровизуализационных изменений, но при наличии характерной клиники.
Характерным (но не специфичным) нейровизуализационным признаком БА является атрофия гиппокампа, которая выявляется на коронарных срезах. Диффузная церебральная атрофия менее значима для диагноза, однако высокий темп атрофического процесса, выявляемый при повторных КТ- или МРТ-исследованиях, также служит дополнительным подтверждением диагноза (см. рис. 2.1). Методы функциональной нейровизуализации (позитронно-эмиссионная томография, однофотонно-эмиссионная компьютерная томография) выявляют снижение метаболизма и кровотока в медиобазальных отделах лобных долей, глубинных и задних отделах височных долей и в теменных долях головного мозга [2, 3, 4, 22, 28]. В последние годы разработан метод прижизненной визуализации бета-амилоида в головном мозге с помощью позитронно-эмиссионной томографии с применением специального радиофармпрепарата, тропного к фрагменту амилоидного белка («питсбургская субстанция», PIB).
Дифференциальный диагноз. БА следует дифференцировать с другими заболеваниями с картиной прогрессирующей деменции.
В первую очередь следует исключить потенциально обратимые виды деменции. К ним относятся дисметаболическая энцефалопатия вследствие соматических и эндокринных заболеваний, дефицитарных состояний (недостаточность витамина В12, фолиевой кислоты), интоксикаций; нормотензивная гидроцефалия, опухоли головного мозга, нейроинфекции. Для выявления указанных состояний все пациенты с деменцией должны пройти полноценное клиническое, лабораторное и инструментальное обследование, включая нейровизуализацию.
Чаще всего проводится дифференциальный диагноз между БА и сосудистой деменцией, другими нейродегенеративными заболеваниями.
Для сосудистой деменции в большинстве случаев характерно преобладание в структуре когнитивных нарушений дизрегуляторных расстройств (нарушение планирования, организации деятельности) при относительно сохранной памяти на события жизни в начале деменции. Другим важным отличительным признаком является наличие уже на стадии легкой деменции выраженной очаговой неврологической симптоматики, прежде всего в виде псевдобульбарного синдрома, нарушений походки. При КТ/МРТ головного мозга выявляются последствия острых нарушений мозгового кровообращения и/или выраженный лейкоареоз, нередко гидроцефалия. В то же время наличие сердечно-сосудистых заболеваний не может служить дифференциально-диагностическим признаком, так как артериальная гипертензия, атеросклероз и сахарный диабет являются факторами риска не только сосудистой деменции, но и БА.
Следует отметить, что не менее чем в 15 % случаев деменции в пожилом возрасте отмечается сосуществование сосудистого поражения головного мозга и альцгеймеровского дегенеративного процесса (так называемая смешанная деменция). В этих случаях в клиническом статусе одновременно отмечаются признаки обоих заболеваний (см. «Сосудистые и смешанные когнитивные расстройства»).
При деменции с тельцами Леви на первый план клинической картины выступают замедленность и заторможенность психических процессов, колебания концентрации внимания (так называемые флюктуации). Другой отличительной особенностью являются повторяющиеся зрительные галлюцинации в виде образов животных или людей. В нейропсихологическом статусе наряду с умеренными нарушениями памяти важное место занимают нарушения зрительно-пространственного гнозиса и праксиса, однако отсутствуют речевые расстройства. Двигательные нарушения представлены различными по выраженности экстрапирамидными расстройствами, такими как гипокинезия, ригидность, постуральная неустойчивость, реже — акционный и/или статический тремор. Также весьма характерна периферическая вегетативная недостаточность. Специфический нейровизуализационный признак — значительное расширение задних рогов боковых желудочков (см. «Деменция с тельцами Леви»).
Лобно-височная дегенерация обычно начинается в пресенильном возрасте (50–65 лет). Характеризуется в первую очередь снижением критики и связанными с этим поведенческими нарушениями: импульсивность, бестактность, пренебрежение принятыми в обществе нормами поведения; изменением пищевого и сексуального поведения. Обычно данные расстройства сочетаются с нарушениями речи по типу акустико-мнестической и/или динамической афазии. В редких случаях заболевание может дебютировать с речевых нарушений (так называемая первичная прогрессирующая афазия). В отличие от БА, память на события жизни, пространственный гнозис и праксис, ориентировка в месте и времени длительное время остаются сохранными. В неврологическом статусе определяются симптомы орального автоматизма, хватательный рефлекс, феномен «противодержания» при исследовании мышечного тонуса, в редких случаях — симптомы паркинсонизма. Специфическим (но не обязательным для диагноза) нейровизуализационным признаком является локальная атрофия лобных и передних отделов височных долей головного мозга, нередко односторонняя (см. «Лобно-височная дегенерация»).
Лечение. Лечение БА должно быть направлено на остановку прогрессирования заболевания (нейропротекторная терапия) и уменьшение выраженности уже имеющихся симптомов.
Возможности нейропротекторной терапии БА остаются на сегодняшний день весьма ограниченными. В экспериментальных работах и в рамках клинических исследований предпринимаются попытки воздействия на основные звенья патогенеза БА, исходя из представления об амилоидном каскаде как ведущем механизме развития данного заболевания [10, 15, 22, 24, 28] (табл. 3.4). Однако в клинической практике данные подходы пока не используются, хотя вероятность их скорого внедрения в практику высока.
Нейропротекторная терапия БА
| Фармакологическая мишень | Терапевтический подход |
|---|---|
| Метаболизм предшественника амилоидного белка | Ингибиторы бета- и гамма-секретаз Активаторы альфа-секретазы |
| Агрегация фрагментов альфа-бета-42 в бета-амилоид | Ингибиторы амилоидогенеза |
| Нейротоксичность бета-амилоида | Активная и пассивная антиамилоидная вакцинация |
| Образование нейрофибриллярных сплетений | Ингибиторы фосфорилирования тау-протеина |
С целью первичной и вторичной профилактики БА оправдано воздействие на модифицируемые факторы риска БА у лиц среднего и пожилого возраста. Целесообразны установление контроля артериальной гипертензии, сахарного диабета, гиперлипидемии, других сосудистых факторов риска, включение в рацион продуктов, богатых природными антиоксидантами (цитрусовые, оливковое масло, красное вино и др.), умеренные умственные и физические нагрузки. Эпидемиологические наблюдения свидетельствуют, что указанные мероприятия уменьшают риск возникновения и темпы нарастания когнитивных нарушений, в том числе у генетически предрасположенных к БА лиц.
Лечение клинически манифестной БА зависит от выраженности когнитивных расстройств. На додементных стадиях (при легких и умеренных нарушениях) используются лекарственные препараты, улучшающие церебральную микроциркуляцию и нейрометаболические процессы. На сегодняшний день имеется положительный опыт применения стандартизованного экстракта гинкго билоба (ЕGb 761), пирацетама в дозах 2,4–4,8 г/сут., ницерголина, пирибедила, фосфатидилхолина, внутривенных вливаний церебролизина, актовегина и др. Остается открытым вопрос о длительности применения указанных препаратов. На сегодняшний день преобладает точка зрения о целесообразности продолжительных курсов (6 мес. и более).
На стадии деменции для уменьшения выраженности основных симптомов БА успешно применяются препараты, оптимизирующие синаптическую передачу. С этой целью используются ингибиторы ацетилхолинэстеразы и антиглутаматные препараты.
Основанием для применения ацетилхолинергических препаратов в терапии БА послужили исследования, в которых была показана корреляционная связь между выраженностью ацетилхолинергической недостаточности и тяжестью когнитивных нарушений и других симптомов БА. Опыт применения ингибиторов ацетилхолинэстеразы в клинической практике подтвердил эффективность данного терапевтического подхода. Показано, что на фоне ацетилхолинергической терапии наблюдается уменьшение выраженности когнитивных и поведенческих нарушений, улучшается адаптация в повседневной жизни, снижается нагрузка на ухаживающих лиц. Противопоказаниями для применения данных препаратов являются синдром слабости синусового узла, брадикардия, тяжелая бронхиальная астма, заболевания печени, почечная недостаточность, неконтролируемая эпилепсия [1, 22, 24].
Схема назначения ингибиторов ацетилхолинэстеразы при БА
| Препарат | Начальная доза | Периодичность увеличения дозы, нед. | На сколько увеличивать, мг/сут. | Максимальная доза |
|---|---|---|---|---|
| Донепезил (арисепт) | 5 мг 1 раз в день | 4 | 5 | 10 мг/сут. |
| Ривастигмин (экселон) | 1,5 мг 2 раза в день | 4 | 3,0 | 6 мг 2 раза в день |
| Галантамин (реминил) | 4 мг 2 раза в день | 4 | 8 | 12 мг 2 раза в день |
| Ипидакрин (нейромидин) | 20 мг 2 раза в день | 2 | 20 | 40 мг 2 раза в день |
В настоящее время для лечения БА применяются 4 ингибитора ацетилхолинэстеразы (табл. 3.5). В начале лечения тем или иным ингибитором ацетилхолинэстеразы в процессе титрования дозы у 10–15 % возникают побочные эффекты в виде головокружения, тошноты, рвоты, диареи или анорексии. Данные побочные явления не угрожают здоровью пациентов и обязательно проходят при уменьшении дозы. Однако в этих случаях не следует стремиться к достижению максимальных доз, но нужно остановиться на хорошо переносимой дозе препарата. В настоящее время активно разрабатываются и внедряются новые лекарственные формы ацетилхолинергических препаратов, более удобные в отношении режима дозирования и с меньшей частотой побочных эффектов. К таким формам относится, в частности, ривастигмин в форме накожного пластыря. Использование накожного пластыря позволяет значительно сократить время титрования дозы и таким образом уменьшить время достижения максимальной эффективности терапии. Одновременно удается минимизировать число холинергических побочных эффектов. Использование пластыря позволяет увеличить приверженность терапии, так как многие пациенты с БА негативно настроены в отношении перорального приема лекарств.
Критерием эффективности ацетилхолинергической терапии является улучшение или стабилизация симптомов на протяжении не менее 6 мес. Если, несмотря на проводимую терапию, продолжается ухудшение когнитивных функций, следует заменить используемый препарат на другой ингибитор ацетилхолинэстеразы.
Помимо ингибиторов ацетилхолинэстеразы в лечении БА используется неконкурентный обратимый антагонист N-метил-D-аспартат-рецепторов к глутамату мемантин. Применение данного препарата уменьшает повреждающее действие глутамата на ацетилхолинергические нейроны и таким образом способствует их большей выживаемости и улучшению состояния ацетилхолинергической системы. Противопоказание к назначению мемантина — неконтролируемая эпилепсия. Препарат, как правило, хорошо переносится. Побочные эффекты в виде возбуждения, нарушений ночного сна крайне редки. Данный препарат назначается в начальной дозе 5 мг 1 раз в день, далее суточная доза увеличивается на 5 мг каждую неделю до терапевтической (20 мг/сут. в 2 приема) [1, 24, 28].
Как ацетилхолинергические препараты, так и мемантин, способствуют регрессу основных симптомов деменции: когнитивных, поведенческих, психотических и функциональных нарушений. Регресс поведенческих и психотических нарушений на фоне базовой терапии во многих случаях позволяет обойтись без применения нейролептиков. В дальнейшем (через 1–2 года и более от начала терапии) выраженность нервно-психических расстройств может увеличиваться, однако менее быстрыми темпами по сравнению с «естественным» течением заболевания.
Ингибиторы ацетилхолинэстеразы и мемантин воздействуют на различные фармакологические мишени и не образуют лекарственного взаимодействия, поэтому могут назначаться одновременно. Наиболее целесообразна комбинированная терапия при недостаточной эффективности монотерапии.
С симптоматической целью при развитии депрессии или поведенческих расстройств применяются антидепрессанты и нейролептики.
Антидепрессанты назначаются при наличии синдрома депрессии, которая нередко развивается в начале БА. Согласно общепринятой гериатрической практике, при сочетании когнитивных нарушений и депрессии терапию следует начинать с лечения депрессии, так как когнитивные расстройства в таком случае могут иметь вторичный характер по отношению к эмоциональным нарушениям. Лечение депрессии проводится по стандартным схемам. Используют препараты без дополнительного холинолитического эффекта, так как последний крайне нежелателен для пожилых лиц с когнитивными расстройствами. Наиболее предпочтительны селективные ингибиторы обратного захвата серотонина и ингибиторы обратного захвата серотонина и норадреналина [22, 28].
При наличии выраженных поведенческих нарушений, не отвечающих на ацетилхолинергическую и/или глутаматергическую терапию, назначаются нейролептики. Показаниям для назначения данного класса препаратов являются бред, галлюцинации, психомоторное возбуждение, агрессивность. Предпочтительны атипичные нейролептики, которые реже вызывают экстрапирамидные побочные эффекты (кветиапин, оланзапин, рисперидон, клозапин). При наличии в неврологическом статусе экстрапирамидных симптомов типичные нейролептики противопоказаны [22, 28].
У пациентов с БА следует воздерживаться от назначения бензодиазепинов и барбитуратов, так как данные лекарственные препараты могут оказывать негативный эффект в отношении когнитивных функций и поведения. Для симптоматического лечения нарушений сна можно использовать препараты мелатонина, низкие дозы зопиклона.
Из нелекарственных методов в лечении пациентов с БА используют упражнения по тренировке памяти и внимания. Они включают в себя обучение специальным приемам, облегчающим запоминание и воспроизведение, и упражнения, направленные на повышение концентрации внимания (см. главу 2). Данные методики наиболее эффективны на стадии умеренных когнитивных нарушений и легкой деменции.
источник