Болезнь Альцгеймера (AD) является основной причиной деменции у пожилых людей, что приводит к потере памяти и снижению познавательной способности. Механизм, лежащий в основе возникновения заболевания, не был полностью выяснен. Однако характерные патологические проявления включают внеклеточное накопление и агрегацию β-пептида амилоида (Aβ) в бляшки и внутриклеточное накопление и агрегацию гиперфосфорилированного тау, образуя нейрофибриллярные клубочки. Несмотря на обширные исследования во всем мире, лечение, модифицирующее болезнь, пока недоступно. В этом обзоре мы фокусируемся на генной терапии как на возможном лечении АД, а также обобщаем недавнюю работу в этой области, начиная от исследований концептуальных концепций на животных моделях и заканчивая клиническими испытаниями. Многофакторные причины AD предлагают множество возможных целей для генной терапии, включая два нейротрофических фактора роста, фактор роста нервов и нейротрофический фактор мозга, ферменты, разрушающие Aβ, такие как неприлизин, эндотелин-конвертирующий фермент и катепсин B и AD ассоциированный аполипопротеин E. В этом обзоре также обсуждаются преимущества и недостатки различных быстро развивающихся вирусных методов доставки генов для генной терапии. Наконец, кратко обобщаются подходы, направленные на снижение уровня белка-предшественника амилоидного белка (APP) и β-сайта APP-расщепляющего фермента 1 посредством опосредованного siRNA нокдауна. В целом, перспективы, похоже, обнадеживают, что генная терапия может стать болезнью, модифицирующей лечение AD.
Болезнь Альцгеймера (AD) является нейродегенеративным заболеванием, приводящим к деменции, которая поражает примерно 29 миллионов человек во всем мире [1]. Поскольку в большинстве стран средняя продолжительность жизни растет, а старение является основным фактором риска развития АД, число пациентов будет продолжать расти. К сожалению, никакое лечение, модифицирующее болезни, доступно сегодня. Болезнь клинически характеризуется прогрессирующим снижением познавательной способности, потерей памяти и изменением личности, сопровождающейся поведенческими и психологическими симптомами деменции, такими как ненормальное поведение, агитация и перепады настроения. Патологические характеристики мозга AD включают амилоидные бляшки и нейрофибриллярные клубочки (NFT), которые образуются путем агрегации внеклеточного амилоидного β-пептида (Aβ) и внутриклеточного гиперфосфорилированного тау, соответственно. Кроме того, наблюдается выраженная потеря синапсов и нейронов, сопровождающаяся увеличением желудочков, и патология связана с клинически наблюдаемым когнитивным снижением, хотя дегенеративные процессы могут начаться существенно до появления первых клинических проявлений.
Aβ образуется из мембранного белка типа I, белка предшественника амилоида (APP) путем последовательных протеолитических расщеплений, выполняемых β-секретазой (β-сайт APP-расщепляющим ферментом 1, BACE1) и γ-секретазой, который состоит из пресенилина (PSEN) , Aph-1, Pen-2 и nicastrin. опосредованное β-секретаса расщепление APP приводит к образованию растворимого N-концевого фрагмента (βAPPs) и связанного с мембраной C-концевого фрагмента (CTF-β), который далее расщепляется γ-секретазой для высвобождения Aβ, который состоит из 40- 43 аминокислоты. APP, который был хорошо сохранен на протяжении всей эволюции, присутствует почти во всех клетках и обильно экспрессируется в нейронных клетках. Его физиологическая роль не была выяснена, хотя некоторые исследования показали, что APP обладает нейропротекторными или нейротрофическими функциями. Гены, кодирующие PSEN1 и PSEN2 и APP, были идентифицированы как причинные в раннем начале семейного AD (FAD). Связанные с FAD мутации в этих генах с аутосомным режимом наследования приводят к агрессивной патологии и увеличивают либо общий уровень Aβ, либо отношение Aβ42 / Aβ40. Aβ42 является более гидрофобным и более склонным к агрегату, чем Aβ40. Эти наблюдения сильно указывают на причинно-следственную связь между накоплением Aβ и патогенезом AD. Однако амилоидные бляшки не обязательно могут быть основной причиной, лежащей в основе гибели нейрональных клеток, синаптической потери и, в конечном итоге, атрофии головного мозга в AD. Имеются данные о том, что растворимые Aβ-олигомеры, а не амилоидные бляшки, являются основными патологическими видами, вызывающими нарушения синаптических и нейронных функций. Например, было показано, что олигомеры Aβ являются высокотоксичными, блокируют долгосрочное потенцирование (LTP), вызывают синаптическую ретракцию и приводят к ухудшению памяти и когнитивных функций [2]. С другой стороны, амилоидные бляшки (фибриллы Aβ) вызывают пролиферацию и активацию глиальных клеток, которые выделяют цитотоксические факторы, и поэтому могут косвенно участвовать в нейронной дисфункции. Следует, однако, отметить, что ни одна из моделей трансгенных мышей, которые накапливают олигомеры Aβ и бляшки Aβ, не воспроизводила нейродегенеративные патологии, поэтому ни одна из гипотез не может считаться экспериментально доказанной. Недавно было показано, что также Aβ43 является компонентом внеклеточных бляшек в мозгу AD и может играть роль в агрегации ([3] и Сайто и Саидо [личное сообщение]). Было высказано предположение, что некоторые рецепторы могут быть вовлечены в связывание Aβ, и недавние исследования показали, что Aβ, связанный с клеточным прионным белком или α7-никотиновым ацетилхолиновым рецептором (α7nAChR), вызывает невропатологию, обеспечивая дополнительную поддержку патофизиологической роли Aβ в AD [4 -6]. В целом имеются значительные свидетельства того, что концентрация, зависящая от концентрации Аβ, является одной из основных причин возникновения АД, хотя, вероятно, другие факторы также могут вносить вклад, в том числе гиперфосфорилированный тау, аполипопротеин Е (АПОЭ) — связанный липидный обмен и воспаление [7-9] ,
Aβ постоянно метаболизируется в головном мозге, а увеличенные стационарные уровни Aβ могут быть вызваны не только повышенным образованием Aβ, но и уменьшением деградации Aβ. Несколько протеаз были вовлечены в деградацию Aβ; neprilysin, инсулин-деградирующий фермент (IDE), катепсин B, матриксные металлопротеиназы, плазмин, эндотелин-конвертирующий фермент (ECE) и ангиотензинпревращающий фермент (ACE) (см. [10, 11]). Среди этих протеаз neprilysin был идентифицирован как основной Aβ-разрушающий фермент [12], в то время как вклад ЕЭК и IDE представляется незначительным [10]. Aβ-разрушающие ферменты могут быть мишенями для лечения, снижающих Аβ, например, в терапевтических подходах генов, направленных на усиление Aβ-деградирующей активности.
Учитывая увеличение числа пациентов с АД, следует учитывать дополнительные затраты на лечение деменции, и не в последнюю очередь, личные страдания пациентов и их опекунов, следует рассмотреть все возможные стратегии лечения. Ингибиторы ацетилхолинэстеразы (AChE), такие как донепезил, ривастигмин и галантамин и антагонист NMDA, мемантин, в настоящее время являются единственными доступными препаратами против деменции. Они временно улучшают симптомы, но, к сожалению, не прекращают прогрессирование заболевания. Однако некоторые перспективные эффекты, модифицирующие болезнь, были достигнуты с помощью иммунотерапии с использованием Aβ-вакцины, и несколько клинических испытаний продолжаются [13]. Кроме того, накопление данных показало, что генная терапия является эффективным способом лечения различных заболеваний и может также быть потенциальным лечением AD.
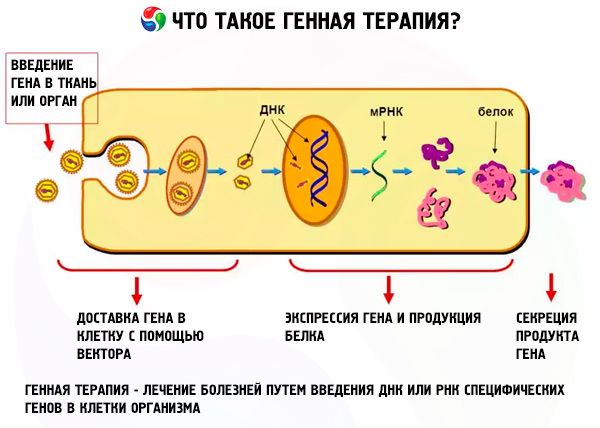
Генная терапия может непосредственно усиливать ферментативную активность и компенсировать снижение уровня биологически активных веществ. Это достигается за счет доставки трансгена, опосредованного вектором, чаще всего вирусом, который инфицирует клетки-хозяева, в которых затем экспрессируется ген. Действительно, генная терапия успешно применяется к нескольким нейродегенеративным, а также неврологическим заболеваниям. На сегодняшний день более 1340 клинических испытаний в 28 странах, нацеленных на более чем 100 генов, были одобрены для целого ряда заболеваний, в основном связанных с раком [14]. Генная терапия временно сталкивалась с проблемой, связанной с безопасностью использования вирусных векторов (см. Таблицу 1 для краткого изложения вирусных векторов, используемых в генной терапии и их свойствах). Например, в клиническом исследовании генной терапии Х-связанного тяжелого комбинированного иммунодефицита с использованием ретровирусов [15, 16] генетический материал из вирусного вектора был вставлен в близость онкогена LMO2, что привело к лейкемии у пяти пациентов, из которых один умер [17-19]. Однако такие случаи редки, и было проведено несколько успешных испытаний. Например, ретровирусная передача, опосредованная вектором гена, была использована для лечения метастатической меланомы и хронического гранулематозного заболевания с использованием передаваемых генам Т-клеток и стволовых клеток крови соответственно [20, 21]. Разработка векторов с использованием онковирусов, в которых гены, необходимые для репликации в неопухолевых клетках, удаляются и где экспрессия гена-мишени управляется опухолевыми / тканеспецифическими промоторами для обеспечения экспрессии генов, специфичных для опухолей, является значительным шагом вперед для лечение на основе генной терапии [22]. Для не опухолевых применений, таких как нейродегенеративные заболевания, включая AD, были разработаны альтернативные вирусные векторы, например. рекомбинантный адено-ассоциированный вирусный вектор (rAAV) и лентивирусный вектор, полученный из ВИЧ. В этих векторах были удалены вирусные гены для саморепликации и, в случае rAAV, включение генетического материала в хромосомную ДНК. Эти векторы способны инфицировать не делящиеся нейронные клетки и, как было показано, являются безопасными, только слабо иммунореактивными, специфическими и обеспечивают долговременное выражение (до 6 лет [23]). В настоящее время проводится несколько клинических испытаний первой фазы генотипа AD, и по крайней мере один из них дал положительные предварительные результаты. В другом нейродегенеративном заболевании болезнь Паркинсона, передача внутричерепного гена из трех генов цели с использованием вектора rAAV показала очень эффективные эффекты, модифицирующие болезнь без побочных эффектов (недавно рассмотренный в [24]). В этом обзоре до настоящего времени обобщаются отчеты об экспериментальной генной терапии на моделях животных AD и клинические испытания генной терапии AD, а также обсуждается потенциал генной терапии как лечения AD. Молекулярные мишени, исследованные до сих пор, можно разделить на четыре категории в соответствии с их физиологической функцией; (i) нейротрофины (НТ), которые поддерживают рост и синаптическую активность нейронов (фактор роста нервов [NGF] и нейротрофический фактор мозга [BDNF]), (ii) ферменты, непосредственно участвующие в деградации Aβ (neprilysin, ECE и катепсин B) (iii) Факторы, связанные с нагрузкой (APOE) и (iv) белки, участвующие в генерации Aβ (BACE1 и APP), которые являются мишенями для опосредуемой siRNA пониженной регуляцией.
Резюме вирусных векторов, используемых в генной терапии
Сокращения: MLV; Вирус лейкемии Moloney, AAV; Адено-ассоциированный вирус, ВПГ; Вирус простого герпеса.
NGF был первым идентифицированным NT (семейство NT также включает BDNF, NT-3 и NT-4/5) и интенсивно изучается. Было показано, что NGF играет важную роль в развитии периферической нервной системы, где она выделяется клетками-мишенями для достижения сбалансированного производства сенсорных и симпатических нейронов. В мозге NGF синтезируется в гиппокампе и коре головного мозга во время развития и взрослого нейрогенеза, когда происходит непрерывная генерация новых нейронов посредством деления нейральных стволовых клеток. Хотя физиологическая роль нейрогенеза гиппокампа неясна, он может участвовать в функциях обучения и памяти (см. [25]). NGF секретируется и транспортируется из гиппокампа в терминалы аксонов базальных кортикальных нейронов переднего мозга (BFCN), где он поддерживает поддержание и рост холинергических нейронов, которые обеспечивают основные иннервации гиппокампа и коры головного мозга. NGF транслируется как предварительный протеин из двух разных вариантов сплайсинга мРНК, что приводит к образованию одного длинного и одного короткого белка-предшественника, которые далее обрабатываются в сети транс-Гольджи. После этого сигнальный пептид удаляется фурином, сигнальной пептидазой пептида, для получения зрелой формы NGF [26]. NGF вместе с другими НТ активирует ряд сигнальных путей клеток, таких как Ras, внеклеточные сигнально-регулируемые киназы (ERK) и связывающий белок cAMP-ответа, путем связывания с тропомиозин-родственной киназой A (TrkA) и р75-рецепторами присутствующих в BFCN. Помимо своих ролей в нейронном росте и поддержании клеток, NGF также влияет на синаптические функции [27, 28]. Кроме того, считается, что proNGF, который является более обильным, чем NGF [29], участвует в передаче апоптотических клеток через связывание с рецептором р75 [30]. Генный нокаут NGF у мышей приводил к сильному воздействию на сенсорные и симпатические ганглии, но на удивление не наблюдалось никаких дефектов BFCN [31]. С другой стороны, инфузия NGF в мышиной модели синдрома Дауна, которая проявляет неудачу в ретроградном транспорте и дегенерации BFCN, может изменить атрофию мозга, указывая на нейротрофную роль NGF [32].
В отличие от BDNF (см. Ниже), ни белок, ни уровни мРНК NGF в мозге AD не уменьшаются, а уровни повышаются в регионах, пораженных AD, таких как гиппокамп и кору головного мозга (рассмотрен в [33] ). Однако экспонирование BFCN снижается как на уровне NGF, так и на уровне TrkA. Эти явления рассматриваются как результат нарушения переноса аксонов NGF в BFCN из-за образования NFT в гиппокампе. Таким образом, NGF накапливается в гиппокампе, а TrkA понижается в BFCN (см. [34]). Сниженный уровень NGF приводит к усадке нейронов и снижению экспрессии ферментов, участвующих в метаболизме ацетилхолина, то есть холин-ацетилтрансферазы (ChAT) и AChE, в головном мозге AD. Снижение этих ферментов в BFCN уже проявляется во время легкой фазы когнитивного нарушения (MCI), фазы, характеризующейся амнезией, без влияния на другие когнитивные функции до такой степени, что повседневная жизнь сдерживается. Примерно у половины пациентов с МСИ развивается АД. Эти результаты привели к развитию ингибиторов AChE для повышения уровней ACh. Это лечение эффективно, но временно улучшает симптомы пациентов с АД в ранней и средней фазах.
В дополнение к нейротрофным свойствам NGF, недавнее исследование показало, что лишение NGF в клетках PC12 активирует амилоидогенный каскад [35], тем самым связывая NGF с Aβ. Стоит отметить увеличение экспрессии proNGF в AD; это может быть основной причиной смерти нейрональных клеток, наблюдаемой в AD, поскольку proNGF индуцирует апоптозную сигнализацию. Кроме того, сверхэкспрессия анти-NGF-антитела в модели трансгенных мышей приводит к дефициту памяти, когнитивным нарушениям и нейродегенерации, сопровождающимся гиперфосфорилированием тау в BFCN. Патологические и поведенческие эффекты могут быть отменены инфузией NGF (см. [36]). Таким образом, дисбаланс между proNGF и NGF и / или между NGF-рецепторами TrkA и p75 в сочетании с нарушенным переносом антерографа NGF, по-видимому, тесно связан с повреждением холинергических нейронов и патологией AD, что указывает на то, что NGF может использоваться в лечении от AD.
Целью лечения NGF является поддержание физиологической концентрации NGF для поддержки холинергических нейронов и повышения их активности в дополнение к уменьшенному уровню ACh в базальном переднем мозге. Администрация NGF предлагает более стойкий эффект, чем ингибиторы AChE, и вмешивается в патогенез AD, чтобы замедлить клиническое ухудшение. Фактически, внутрижелудочковая инфузия NGF предотвращала гибель холинергических нейронов, вызванную поражением, и улучшала функции обучения и памяти у старых крыс [32, 37]. Аналогичные результаты были получены у приматов, где инфузия NGF спасла холинергические нейроны от смерти после поражения [38, 39]. Однако для лечения NGF потребовалось бы, чтобы на клеточной поверхности нейронов присутствовало достаточное количество рецепторов TrkA и р75, и поэтому совместное предоставление этих рецепторных генов с геном ChAT могло бы быть предпочтительным для получения максимального эффекта.
После этих многообещающих результатов было начато первое клиническое испытание первой фазы с NGF у людей [40, 41]. Поскольку NGF слишком велик, чтобы пересечь гематоэнцефалический барьер (BBB), он вводился внутрижелудочно пациентам AD. Тем не менее, NGF-инфузия активировала сенсорные нейроны в ноцицептивном ответе, что приводило к потере веса и тяжелой боли в спине, и поэтому испытание было остановлено [42]. Аналогичным образом, инфузия NGF у животных приводила к потере веса, прорастанию аксонов и миграции клеток Шванна в продолговатый мозг и спинной мозг [36]. Эти результаты выявили недостаток внутрижелудочкового введения, то есть широкое и неконтролируемое распределение по всему мозгу приводило к неожиданным неблагоприятным последствиям. С другой стороны, исследование показало некоторое улучшение когнитивной функции и наблюдалась повышенная активность нейронов у пациентов.
Первое клиническое исследование с прямой инфузией NGF продемонстрировало необходимость методов, которые обеспечивают лучший контроль над распределением и уровнем NGF в головном мозге, чтобы избежать возникновения ноцицептивного ответа или других побочных эффектов. Генная терапия с использованием подходов ex vivo с приживлением NGF-экспрессирующих клеток или подходов in vivo с прямой инъекцией вектора, несущего ген NGF, предлагает такие возможности. Подходы ex vivo, например, с использованием фибробластов, трансдуцированных ретровирусными векторами, такими как вирус лейкемии Moloney (MLV), были успешно применены как у грызунов, так и у обезьян и показали, что они имеют долгосрочное выражение, по крайней мере, до 1 года. Как поврежденные поражением холинергические нейроны, так и связанные с возрастом атрофии головного мозга были отменены без неблагоприятных аффектов, а когнитивные функции были улучшены ([43-50] и таблица 2). Таким образом, было начато первое клиническое исследование I фазы генной терапии, в которой участвовали восемь пациентов с АД, с использованием вектора MLV для экспрессии NGF [51]. Фибробласты кожи, трансдуцированные MLV-вектором, несущим ген NGF для экспрессии NGF (50-75 нг / 10E6 клеток), вводили в базальную базу пациентов. Когнитивные функции были протестированы, а активность нейронов контролировалась с помощью позитронно-эмиссионной томографии, и после 2-летнего периода наблюдались значительные улучшения как в метаболизме глюкозы, так и в познании без видимых побочных эффектов. К сожалению, у одного пациента развились кровоизлияния из-за движения во время инъекции и в конечном итоге умер. Анализ после вскрытия выявил значительный рост холинергических нервов вокруг места инъекции. Это было первое прямое наблюдение у человека, показывающее нейротрофический эффект NGF. В совокупности клиническое исследование показало, что локальное приживление фибробластов, экспрессирующих NGF в пораженных областях мозга AD, оказывает нейротрофическое воздействие на холинергическую систему, что приводит к улучшению нарушенной когнитивной функции без прямого отрицательного воздействия. Наконец, в 2008 году на Karolinska Intitutet была начата клиническая фаза 1b исследования на основе инкапсулированных эпителиальных клеток из сетчатки, сконструированной для экспрессии NGF [52]. В исследовании приняли участие шесть пациентов, получивших полупроницаемый имплантат биодедущей, который позволил бы высвободить NGF и предотвратить миграцию клеток без осложнений после операции.
Резюме исследований генной терапии в AD
Сокращения: B.F .; базальный передний мозг, N.B .; Ядро базалис, N.B.M .; базальная база ядра Meynert, F.C .; лобная кора, MLV; Вирус лейкемии Moloney, rAAV; рекомбинантный адено-ассоциированный вирус, siRNA; короткая мешающая РНК.
Поскольку доставка гена ex vivo требует трудоемкой клеточной препаративной работы, менее обширная доставка гена in vivo будет иметь значительные преимущества. Кроме того, исследования доставки генов первого поколения были основаны на трансплантатах клеток, трансдуцированных ретровирусными вирусами, например. MLV. Хотя ретровирусы были некомпетентными и нетоксичными, возможный онкогенный риск говорит в пользу использования других вирусных векторов, таких как вирусы AAV, которые были разработаны для приложений доставки гена in vivo (см. Таблицу 1 для краткого изложения используемых вирусных векторов в генной терапии и их свойствах). AAV, который способен инфицировать как делящиеся, так и не делящиеся ячейки, например. нейронов, является перспективным вектором генной терапии в центральной нервной системе (ЦНС). Обширные исследования безопасности показали, что AAV нетоксичен, слабо иммунореактивен и невосприимчив, и он может быть дополнительно спроектирован так, чтобы быть нерепликативным и иметь очень низкую вероятность интеграции в хромосому из ее эпизодической стадии. Доклинические исследования у мышей показали, что доставка гена NGF, опосредованного rAAV, может вызывать стабильную экспрессию NGF, которая улучшает вызванную поражением холинэргическую дегенерацию и отменяет когнитивный дефицит у старых мышей [53-57]. Существует несколько серотипов rAAV, и в зависимости от белков, отображаемых на поверхности их капсидов, они будут связываться с различными рецепторами клеточной поверхности, что приводит к различной аффинности для разных типов клеток. Следовательно, серотипы могут быть использованы для определения специфичности трансдукции. Клетки в ЦНС эффективно трансдуцированы серотипами rAAV 1, 2 и 4-9 с дифференциальным тропизмом и токсичностью [58, 59]. Большинство исследований по доставке генов в ЦНС проводили с использованием AAV-2 и 5, а серотип 5 приводил к более широко распространенной инфекции нейронов и астроцитов по сравнению с нейроноспецифическим серотипом 2. Промотором цитомегаловируса (ЦМВ) является наиболее широко используемый промотор для стимулирования экспрессии трансгена, хотя использовали другие промоторы, специфичные для клеточного типа, такие как нейроспецифический промотор энолазы. Кроме того, включение регулирующих элементов, например. Пост-транскрипционный элемент вируса гепатита Woodchuck увеличивает частоту трансдукции. Альтернативой AAV является вирусный вектор чечевицы, полученный из ВИЧ; однако, хотя этот вектор был спроектирован для предотвращения репликации и сборки капсидов, его происхождение ВИЧ вместе с теоретическим онкогенным риском при хромосомной инкорпорации привело к некоторому нежеланию использовать его в клинических испытаниях. С другой стороны, хромосомная интеграция, по-видимому, не является серьезным риском, поскольку значительное большинство нейронов не делятся (исключение нейронных стволовых клеток), а развитие дефицитного по степени интеграции лентивируса еще больше повысит безопасность. Продолжающееся исследование механизмов вирусной инфекции, вероятно, увеличит знания и полезность rAAV и лентивирусов. Многообещающие доклинические результаты доставки гена NGF с использованием rAAV и данные клинических испытаний доставки гена NGF ex vivo, а также rAAV-опосредованная генная терапия при таких заболеваниях, как PD и рак (обзор в [60]), побудили исследователей инициировать клиническое исследование, основанное на rAAV-опосредованной генной терапии NGF [61].
BDNF — относительно небольшой (14 кД), гомодимерный белок, который был хорошо сохранен на протяжении всей эволюции и относится к семейству NT. Хорошо известно, что BDNF, который имеет 50% идентичность последовательности с NGF, участвует в развитии, поддержании и развитии нервных клеток как в ЦНС, так и в периферической нервной системе. BDNF высоко экспрессируется во всем мозге, включая регионы, имеющие важное значение для когнитивных и интеллектуальных функций, таких как гиппокамп и кору головного мозга [62]. BDNF также экспрессируется в клетках вне мозга, например. в эндотелиальных и гладкомышечных клетках [63], где считается, что его экспрессия поддерживает периферические нервные клетки. Было идентифицировано шесть различных транскриптов гена BDNF, и несколько промоторов обеспечивают тканеспецифическую экспрессию [64, 65]. BDNF локализуется главным образом в сотовой клетке и дендритах, к которой она транспортируется секреторными и послебольшевыми везикулами. МДНК BDNF была обнаружена в дендритах, что указывает на то, что происходит локальный перевод. BDNF связывается с высоким сродством к TrkB-рецептору, который транслируется из двух разных вариантов сплайсинга мРНК, одна из которых представляет собой полноразмерную форму с киназной активностью, тогда как другая представляет собой укороченную и киназную активность-дефицитную форму с низким сродством к рецептор р75 [66, 67]. Интересно, что большое количество BDNF обнаруживается в тромбоцитах крови, где оно может служить в качестве резервуара BDNF для поддержки периферических нейронов. Важно отметить, что BDNF участвует в синаптической пластичности (см. [27]).
Уровень BDNF в головном мозге снижается не только в AD [68-73], но также в PD [74], болезни Хантингтона [75], депрессии [76, 77] и шизофрении [78]. Таким образом, кажется, что препараты на основе BDNF могут найти применение для широкого спектра заболеваний мозга. Интересно отметить, что уровни BDNF увеличиваются посредством физических упражнений и когнитивной стимуляции и снижаются при старении у здоровых людей [79], предполагая, что BDNF может быть связан с когнитивными функциями.
источник
Генетическое лечение, которое доставляет вирус гена в мозге, можно использовать для лечения болезни Альцгеймера на ранней стадии, говорится в исследованиях, опубликованных в журнале.

Болезнь Альцгеймера является наиболее распространенным видом деменции. Это затрагивает более 40 миллионов человек во всем мире, что приводит к потере памяти, замешательству, изменению личности или настроению. В настоящее время нет лекарств.
В 2013 году Центры по контролю и профилактике заболеваний (CDC) оценили, что до 5 миллионов человек жили с болезнью Альцгеймера в Соединенных Штатах. В 2014 году 93 541 смерть была приписана этой болезни, что сделало ее шестой основной причиной смерти в США.
Ученые из Имперского колледжа Лондона в Соединенном Королевстве теперь использовали модифицированный вирус для доставки гена, известного как PGC1-альфа, в клетки мозга мышей. Они обнаружили, что это уменьшает развитие болезни Альцгеймера.
Вирус называется лентивирусным вектором, и он обычно используется в генной терапии.
На основе предыдущих лабораторных исследований команда предсказала, что она может остановить белок, называемый амилоид-бета-пептид, от образования в клетках.
Амилоидные бляшки — это липкие глыбы белка, которые встречаются в мозге людей с болезнью Альцгеймера. Считается, что они ответственны за смерть клеток мозга. Амилоид-бета-пептид является основным компонентом этих бляшек.
Профессор Николас Мазаракис, соавтор исследования, объясняет, как ученые могут повернуть путь к тому, как лентивирус заражает клетки в свою пользу; он включает в себя создание модифицированной версии вируса и использование его для доставки генов в конкретные клетки.
Ученые уже используют эту технику для исследования лечения артрита, рака и других состояний. В клинических испытаниях нынешняя команда успешно использовала его для доставки генов в мозг людей с болезнью Паркинсона.
В этом последнем исследовании исследователи ввели вирус в две области мозга: кору и соседний гиппокамп.
Считается, что болезнь Альцгеймера начинается в коре, а затем постепенно распространяется на гиппокамп. Первый урон может произойти за 10-20 лет до того, как болезнь станет внешне видимой.
Кора ассоциируется с долговременной памятью, рассуждениями, мышлением и настроением. Ущерб может привести к депрессии и затруднению выяснения того, как выполнять знакомые задания.
Гиппокамп играет важную роль в обучении, в преобразовании кратковременных воспоминаний в долгосрочные воспоминания и в ориентации. Повреждение гиппокампа может заставить человека забыть о последних событиях, таких как то, что они сделали сегодня утром. Это также причина, по которой люди с болезнью Альцгеймера теряются на знакомых маршрутах — например, не могут найти дорогу домой.
У мышей, получавших лечение, были ранние стадии болезни Альцгеймера. Они еще не разработали амилоидные бляшки. Им вводили адаптированный вирус, содержащий ген PGC1-альфа.
Через четыре месяца после инъекций тесты показали, что мыши, получившие ген, имели очень мало амилоидных бляшек, лучшую память и отсутствие клеток мозга в гиппокампе. Те, кто не лечился, имели в своем мозгу несколько бляшек.
Чтобы проверить память, команда заменила знакомый объект в клетках мыши новым элементом. Те, у кого здоровая память, исследовали новый объект дольше. Обработанные мыши выполняли также здоровые мыши.
Обработанные мыши также имели меньшее количество глиальных клеток. При болезни Альцгеймера глиальные клетки, как полагают, вызывают дополнительное повреждение клеток, выделяя токсичные воспалительные вещества.
«Хотя эти результаты очень ранние, они предполагают, что эта генная терапия может иметь потенциальное терапевтическое применение для пациентов. Существует много препятствий для преодоления, и в настоящий момент единственным способом доставки гена является инъекция непосредственно в мозг, это доказательство концептуального исследования показывает, что этот подход требует дальнейшего изучения ».
Старший автор доктор Магдалена Састр
Авторы надеются, что полученные результаты могут открыть новый путь к будущей терапии, чтобы предотвратить болезнь или остановить ее на ранних стадиях. Они надеются начать изучать, как применять лечение у людей, хотя они отмечают, что за несколько лет до того, как его можно будет использовать в клинических условиях.
Они считают, что это лечение лучше всего использовать на ранних стадиях болезни Альцгеймера, когда появляются симптомы.
Доктор Дэвид Рейнольдс, главный научный сотрудник Исследовательского института Альцгеймера в Великобритании, называет результаты «многообещающим шагом на пути к разработке методов лечения этого разрушительного состояния».
PGC1-альфа играет роль в регуляции обмена сахара и жира в организме. Другие исследования показали, что физические упражнения и ресвератрол — соединение, которое присутствует в красном вине — могут увеличить уровни PGC1-альфа.
Узнайте, как успех в тестах словесной памяти может означать, что у некоторых женщин отсутствует ранняя диагностика болезни Альцгеймера.
источник
Болезнь Альцгеймера остается одной из самых непонятных для врачей. На данный момент для объяснения возможных причин этого заболевания были предложены три основные конкурирующие гипотезы. Но есть один факт о недуге, который приобрел почти неопровержимый статус. Именно его и собираются использовать ученые, чтобы разработать терапию, предотвращающую болезнь Альцгеймера.
Этот факт касается гена APOE, который также называют «геном забывчивости». У него есть три версии (2,3 и 4). Если вы унаследовали версию 2, то риск развития заболевания головного мозга может быть в два раза ниже среднего. Версия 3 имеет средние показатели. А вот 4 – радикально увеличивает риск (до 12 раз).

Даже врачи пытаются избегать анализов на APOE, так как плохой результат в чем-то сродни приговору. Лечения не существует, а гены изменить нельзя… Или все-таки можно?
Вероятно, что в скором времени ученые найдут способ. Врачи из Нью-Йорка начнут тестировать новую генную терапию с мая этого года. Исследователи собираются давать людям с самыми «неудачными» генами APOE большую дозу версии, которая понижает риск.
Ученые надеются, что это поможет замедлить развитие болезни у пациентов с Альцгеймером. Положительный результат в свою очередь открывает перспективу предотвращать недуг на этапе выявления «нехорошего» гена. Руководитель клинических испытаний – Рональд Кристал из колледжа Уэилл Корнелл Медикал в Манхэттене. Это не только новая тактика против деменции, но и инновация в генной терапии. Сейчас большая часть действий по замене генов полагается на вирусы, которые переносят инструкции ДНК в человеческие клетки. Такая замена одного «неисправного» гена направлена на устранение редких заболеваний, например, гемофилии. Однако у более распространенных болезней нет подобных одиночных причин. И именно поэтому генную терапию не называют особенно многообещающей.
По словам Кирана Мусунуру, который является профессором медицинской школы Пенсильванского университета, очень похоже на то, что путь к клиническим испытаниям на людях будет длительным. «Тем не менее, существует острая необходимость в хоть каком-либо лечении», – добавляет ученый. Мусунуру занимается изучением генетических методов лечения сердечнососудистых заболеваний. Ученый утверждает, что запланированный в колледже Уэилл Корнелл Медикал эксперимент представляет собой новый вид генной терапии. Ведь его цель – не вылечить, а снизить риск возникновения заболевания.
Рональд Кристал признается, что план его исследования также обходит стороной споры о причинах болезни Альцгеймера. Эта дискуссия давно уже стала «полем чудес с многомиллиардным оборотом, где проигрывают и пациенты, и фармацевтические компании». Так, в январе одна из них – Roche остановила два масштабных исследования антител. Их задачей было выяснение свойств бета-амилоидных бляшек. По одной из теорий, именно эти бляшки вокруг нейронов и приводят к развитию Альцгеймера.
«Много тех, кто уверен: виной всему амилоид», – рассказывает Кристал. По иной версии, виноват тау – другой белок, клубки которого были обнаружены в умирающих нейронах. «Вероятно, найти ответ будет сложно. Но мы избрали подход, который игнорирует все это, рассматривая ситуацию именно с генетической точки зрения», – добавляет ученый.
Команда исследователей собирается обратиться к открытию 25-летней давности. Так, в 1990-х годах в Университета Дьюка искали белки, которые могли крепиться к амилоидным бляшкам, в результате чего был выявлен аполипопротеин-е, кодирующийся геном APOE. Ученые сделали секвенирование генов у более чем 100 пациентов с Альцгеймером и обнаружили: отдельная версия (APOE4) была распространена поразительным образом среди больных.
Функция данного гена не до конца понятна. APOE играет роль в транспорте жиров и холестерина, и его статус фактора риска также вполне стабилен. Так, Ассоциация Альцгеймера предоставила данные, по которым у примерно 65 % людей с болезнью Альцгеймера есть, по крайней мере, одна копия опасного гена. А для тех, кто родился с двумя копиями высокого риска (по одной от каждого родителя), деменция в старости становится практически гарантированной.
Некоторые люди наследуют одну 2 и одну 4 версии гена. Тогда у них показатели риска ближе к среднему. Предположительно, защитная версия APOE компенсирует рискованную. Врачи Weill Cornell решили скопировать именно этот эффект. Сейчас центр ищет для эксперимента людей с двумя копиями гена высокого риска – тех, кто уже потерял память или болеет Альцгеймером. Затем, по словам Рональда Кристала, первые добровольцы получат в спинной мозг инфузию из миллиарда вирусов, которые несут APOE2.
Ученые основываются на экспериментах с обезьянами – вирусы должны распространить «счастливый ген» по всему мозгу пациента. Кстати, также лечили мышей, в результате чего они накопили меньше амилоида. «Концепция рациональна, но сработает ли она с людьми – это иной вопрос», – добавляет Кристал.
Исследование американских ученых будет предварительным. Рональд Кристал замечает: его команде предстоит определить, функционирует ли добавленный ген на уровне, где можно будет его обнаружить. К сожалению, к моменту, когда человек начинает путать имена и забывать, где лежат ключи от автомобиля, изменения в его мозге происходят уже с десяток лет. А это значит, что те пациенты, которые присоединяются к эксперименту, не могут рассчитывать на многое.
И все-таки, несмотря на это, Фонд поиска лекарства от Альцгеймера дал три миллиона долларов на предстоящее исследование. Ведь оно дает надежду, что люди среднего возраста, которые находятся в группе риска, смогут подвергаться одноразовой генетической настройке. И небольшое снижение скорости происходящих изменений в головном мозге со временем может привести к глобальному решению проблемы.
источник
Никто не знает наверняка, что приводит к болезни Альцгеймера. Но один факт об этой болезни приобрел практически неопровержимый статус. В зависимости от того, какие версии гена APOE вы унаследуете, риск заболевания головного мозга может быть в два раза ниже среднего — или в 12 раз выше. APOE иногда называют «геном забывчивости», и у него бывает три версии: 2, 3 и 4. Версия 2 снижает риск для человека; 3 — средний показатель; 4 — увеличивает риск радикально.

Риск настолько велик, что врачи избегают проверки состояния APOE, поскольку плохой результат может расстроить человека — и с этим ничего не поделать. Лечения нет, а гены изменить нельзя.
Пока нельзя. Но врачи из Нью-Йорка говорят, что, начиная с мая, они начнут тестировать новую генную терапию, в которой людям с самыми неудачными генами APOE будут давать огромную дозу версии, понижающей риск.
Если это поможет замедлить медленное истощение мозга болезнью людям, у которых уже есть болезнь Альцгеймера, в конечном счете это приведет к возможности предотвращения болезни. Клинические испытания, которыми руководил Рональд Кристал из Weill Cornell Medicine в Манхэттене, представляют собой новую тактику против деменции, а также новый поворот в генной терапии. Большинство усилий по замене генов, которые полагаются на вирусы, переносящие инструкции ДНК в клетки человека, направлены на устранение редких заболеваний, таких как гемофилия, путем замены одного неисправного гена.
Но у распространенных заболеваний нет таких одиночных причин, поэтому генная терапия никогда не была особо многообещающей. Торговая группа Alliance for Regenerative Medicine утверждает, что в настоящее время никаких генных терапий не проводится на пациентах с Альцгеймером.
«Похоже на то, что путь к клиническим испытаниям на людях будет долгим, однако есть острая необходимость в любом лечении», говорит Киран Мусунуру, профессор медицинской школы Пенсильванского университета. Он изучает генетические методы лечения сердечно-сосудистых заболеваний и говорит, что эксперимент, запланированный в Нью-Йорке, представляет новую категорию генной терапии, цель которой не в том, чтобы вылечить, а в том, чтобы снизить риск возникновения будущего заболевания у здоровых людей.
Кристал говорит, что его план также обходит стороной дискуссию об истинной причине болезни Альцгеймера, которая превратилась в поле чудес с многомиллиардным оборотом, в котором проигрывают и фармацевтические компании, и пациенты. В январе Roche остановила два больших исследования антител, которые должны были прояснить свойства бета-амилоидных бляшек, последнюю из теорий, которая утверждала, что эти бляшки вокруг нейронов приводят к появлению Альцгеймера.
«В области есть много тех, кто твердо верит, что виной всему амилоид», говорит Кристал. Другие считают, что виноват другой белок — тау — клубки которого нашли в умирающих нейронах. «Ответ, вероятно, будет сложно найти. Подход, который мы избрали, игнорирует все это и рассматривает ситуацию с генетической точки зрения».
При этом команда Кристала полагается на 25-летнее открытие. В 1990-х годах ученые из Университета Дьюка занимались поиском белков, которые могли крепиться к амилоидным бляшкам. И они выявили аполипопротеин-е, который кодируется геном APOE. Секвенируя этот ген у 121 пациента, они обнаружили, что отдельная версия — APOE4 — необъяснимым образом была распространена среди страдающих от этой болезни.
Функция этого гена до сих пор не до конца понятна (он играет роль в транспорте холестерина и жиров), но его статус фактора риска остается пугающим. По данным Ассоциации Альцгеймера, около 65% людей с болезнью Альцгеймера имеют, по крайней мере, одну копию опасного гена. Для людей, родившихся с двумя копиями высокого риска, по одной от каждого родителя, деменция становится почти гарантированной, если они проживут достаточно долго.
Тем не менее, некоторые люди наследуют одну 4 и одну 2, версию гена с низким риском. Эти люди имеют более близкий к среднему риск, что предполагает, что защитная версия гена компенсирует рискованную.
Именно этот эффект врачи Weill Cornell попытаются скопировать. В настоящее время центр ищет людей с двумя копиями гена высокого риска, который уже потеряли память или даже обзавелись Альцгеймером. По словам Кристала, примерно через месяц первые добровольцы получат инфузию в спинной мозг из миллиарда вирусов, несущих ген 2.
Основываясь на тестах на обезьянах, Кристал ожидает, что вирусы распространят «счастливый ген» в клетках по всему мозгу пациента. Мышей лечили точно так же, и грызуны накопили меньше амилоида в мозгах.
Эта стратегия, по мнению исследователя, не зависит от знания всего о том, что действительно вызывает болезнь. «В Альцгеймере нас привлекает очевидная генетическая эпидемиология», говорит он. «Итак, стратегия в том, можем ли мы искупать мозг в E2? У нас есть для этого инфраструктура, поэтому мы подумали, почему бы и нет? Это решает проблему механизма заболевания».
«Концепция рациональна», добавляет Кристал. «Сработает ли это с человеком — другой вопрос».
Нью-Йоркское исследование будет предварительным. Кристал говорит, что его команде нужно определить, функционирует и добавленный ген на уровне, на котором его можно будет обнаружить. Врачи забирают у пациентов спинномозговую жидкость и проверяют, содержит ли она ожидаемую смесь белков — ожидаемый тип 4, но теперь с равным или большим количеством замешанного 2.
К тому времени, когда люди начинают забывать имена и где ключи от машины, изменения в мозге проходят уже десяток лет. Это значит, что пациенты, которые присоединяются к исследованию, могут рассчитывать не на многое. Для них будет уже слишком поздно.
Несмотря на это, Фонд поиска лекарства от Альцгеймера выдал Кристалу 3 миллиона долларов на исследование. В конце концов, есть надежда, что люди среднего возраста с генами в зоне риска смогут подвергаться однократной генетической настройке. Даже небольшое снижение скорости, с которой происходят изменения в мозге, может со временем изменить ситуацию.
Что ж, будем надеяться, что все получится. Предлагаем следить за новостями на нашем новостном канале.
источник

Генная терапия с применением вирусного вектора, доставляющего ген человеческого фактора роста нервов (NGF) к базальному ядру Мейнерта, продемонстрировала высокую безопасность у пациентов с болезнью Альцгеймера легкой и средней тяжести.
Таковы результаты первого испытания инновационной терапии на людях, о которых сообщается на Конференции по клиническим исследованиям болезни Альцгеймера в Сан-Диего.
«Нами не было обнаружено никаких недостатков по сравнению с уже существующими методами терапии нарушений памяти и мышления у пациентов с болезнью Альцгеймера», — заявил на конференции доктор Raymond T. Bartus. Доступные сегодня ингибиторы холинэстеразы, по мнению ученого, «являются неселективными, и их применение во многом ограничивают дозозависимые побочные эффекты».
Доктор Bartus, он же президент базирующейся в Сан-Диего компании RTBioconsultants, сказал: «Их влияние на холинэргическую систему не оптимально, они не восстанавливают нервные клетки, не защищают нейроны от клеточной смерти и не влияют на утраченную нейрональную функцию. Конечно, препараты дают некоторое улучшение, но уровень их эффективности низкий, и они не улучшают симптоматику у всех пациентов. Более того, они никак не влияют на дальнейшее прогрессирование болезни Альцгеймера».
Нейротропная терапия фактором роста нервов должна преодолеть все эти недостатки и дать врачам более мощное средство, чем нынешняя медикаментозная терапия. Десятки лет исследований на животных моделях позволяют предположить, что фактор роста нервов «обладает выраженными антиапоптозными и репаративными свойствами по отношению к некоторым нейронам дегенерирующего головного мозга».
Доктор Bartus сказал: «Есть как минимум два существенных препятствия на пути внедрения этой технологии. Во-первых, болезнь Альцгеймера чрезвычайно сложна, поэтому определение мишеней терапии играет очень большую роль. Правильный выбор холинергических нейронов-мишеней – это настоящий вызов, поскольку именно их дегенерация является главной причиной потери памяти».
Вторым препятствием, по словам доктора, является необходимость воздействия на холинергические нейроны базального ядра Мейнерта, избегая при этом воздействия на другие популяции нейронов. 20 лет назад на практике такое было очень трудно осуществить. В прошлом десятилетии генная терапия в сочетании со стереотаксической хирургией позволила проводить лечение.
Используя новые технологии, доктор Bartus и его коллеги из Ceregene, где исследователь работал главным научным сотрудником более 10 лет, разработали вирусный вектор, который доставляет в клетки человеческий фактор роста нервов (AAV2-NGF, или CERE-110).
«Вы можете представить это как биофармацевтический препарат, вызывающий при введении пациенту экспрессию гена NGF в нейронах-мишенях, и поддерживающий таким способом холинергические нервные клетки. Доклинические исследования продемонстрировали строгое дозозависимое действие на нейроны-мишени, отсутствие побочных эффектов и токсичности, даже после применения у животных очень больших доз. Это было довольно необычно, и очень обнадежило нас», — прокомментировал результаты своих исследований доктор.
Доктор Bartus на этой конференции презентовал результаты клинического исследования фазы I: данные по 10 пациентам в возрасте от 50 до 79 лет с болезнью Альцгеймера легкой или средней тяжести, которые получали три разные дозы AAV2-NGF посредством билатеральной стереотаксической инъекции в базальное ядро Мейнерта. Доза А составляла 1,2×1010 вирусных частиц, доза В 5,8×1010 вирусных частиц, а доза С — 1,2×1011.
Пациентов наблюдали на протяжении 24 месяцев, изучая безопасность и предварительно оценивая эффективность генной терапии. Для этого использовали позитронно-эмиссионное сканирование с 18-фтордезоксиглюкозой перед началом исследования, а затем после 6, 12 и 24 месяцев. Также использовались нейрофизиологические тесты, которые включали шкалу когнитивной оценки при болезни Альцгеймера ADAS-Cog. Компьютерное программное обеспечение давало возможность построить детальные 3D модели базального ядра Мейнерта «с целью изучить распределение CERE-110 в ядре и рассчитать оптимальную дозу NGF».
Доктор Bartus сообщил, что AAV2-NGF безопасен и хорошо переносится на протяжении всех 24 месяцев лечения. Побочные явления, которые были зарегистрированы, связаны с самой хирургической процедурой, и проходили вскоре после проведения (головная боль).
Позитронно-эмиссионная томография не выявила признаков ускорения дегенерации нейронов, вскрытие трех пациентов (умерших в силу разных причин за эти 2 года) подтвердило активную ген-опосредованную экспрессию NGF непосредственно в базальном ядре Мейнерта.
Ученые наблюдали некоторое ухудшение показателей по ADAS-Cog и другим нейрофизиологическим тестам. «Нет никаких клинических свидетельств того, что когнитивное ухудшение у пациентов ускорялось, но мы не можем пока говорить о том, насколько улучшилось их состояние», — сказал доктор.
Эти результаты дают основания для проведения клинического исследования фазы II, которое будет финансироваться Национальным институтом здоровья США как одно из важнейших исследований в области лечения болезни Альцгеймера. Итоги исследования ожидаются не ранее 2015 года.
источник
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Новейший вид лечения – генная терапия – уже продемонстрировала положительные результаты у пациентов с заболеваниями Паркинсона и Гентингтона. Как сообщила газета Таймс, новая методика позволит медицинским специалистам побороть многие тяжелые неврологические патологии, среди которых и такое опасное заболевание, как синдром Альцгеймера – одна из наиболее распространенных разновидностей деменции.
Не так давно в Лондоне проходила очередная неврологическая конференция, на которой активно обсуждалось лечение таких заболеваний головного мозга, как болезнь Паркинсона, Гентингтона и Альцгеймера. При этом ученые настаивали на том, что при помощи генной терапии в скором времени представится возможность не только лечить подобные заболевания, но и осуществлять их профилактику.
Сущность новейшей методики состоит в том, что в определенные зоны головного мозга, которые обладают наибольшей чувствительностью к болезненным нарушениям, вводят элементы вирусов, несущих копии нормальных генов. После этого вирус переносит обновленную генетическую кодировку в клеточные мозговые структуры – в итоге их функции изменяются, тормозится выработка токсичного протеина, высокая концентрация которого провоцирует развитие болезни Альцгеймера.
«На данный момент мы находимся ещё в самом начале пути. Однако уже сейчас мы вполне можем практиковать новую методику лечения. В первую очередь, нас воодушевляет тот факт, что нам удалось применить микровирусы для генной транспортировки в головной мозг», — прокомментировал свое выступление на конференции руководитель биотехнологической корпорации Стивен Пол. Также он заметил, что микровирусы могут деактивировать внутри мозга отдельные гены. «Мы имеем доступ к протеиновым вирусным оболочкам, способным проникать сквозь гематоэнцефалическую мембрану в сотни раз легче, чем известные ранее образцы. И это очень важный момент», — подвел итоги Стивен Пол.
В то же время группа специалистов, представляющих Имперский колледж в Лондоне, заявила о существенных успехах в использовании генной терапии для борьбы со схожими патологиями у грызунов. Во время эксперимента в мозговые структуры мышей с микровирусом был отправлен специфический ген, который положительно повлиял на динамику заболевания.
Болезнь Альцгеймера относят к важнейшей в социальном плане патологии человека. Исследователи и ученые прилагают огромные усилия, чтобы разработать более эффективные методы лечения и предупреждения данного заболевания. Однако до настоящего времени терапевтическая схема ограничивалась в основном симптоматическими препаратами и процедурами, так как утверждалось, что полностью вылечить заболевание практически невозможно.
В настоящее время полученные учеными результаты по данному вопросу ещё не внедрены в медицинскую практику. В то же время результаты многочисленных исследований вселяют определенную надежду. По предварительной оценке специалистов, генная терапия может быть применена на практике уже в самое ближайшее время.
 [1], [2], [3], [4], [5]
[1], [2], [3], [4], [5]
источник
МОЛЕКУЛЯРНЫЕ МЕХАНИЗМЫ БОЛЕЗНИ АЛЬЦГЕЙМЕРА: ГЕНЕТИЧЕСКИЙ ПОДХОД
Е. И. Рогаев Научный центр психического здоровья РАМН, Москва
Центральной задачей при исследовании патогенеза сложного заболевания является идентификация первичных молекулярных нарушений.
Предполагается, что первые гистопатологические изменения (например, накопление амилоидных бляшек) при болезни Альцгеймера могут возникать задолго (за 10-20 лет) до появления первых клинических симптомов. Это, разумеется, затрудняет идентификацию первичных дефектов, ведущих к нейродегенерации. Прорывом в данной области явился генетический подход, в частности, стратегия позиционного клонирования генов.
Генетическая предрасположенность и возраст являются наиболее четко определенными факторами риска развития болезни Альцгеймера (БА).
Установлено, что болезнь Альцгеймера включает несколько генетически гетерогенных форм, объединенных общим спектром клинических и гистопатологических признаков. Обнаружено, что причиной или фактором риска развития определенных форм БА являются мутации и полиморфизм в некоторых генах человека. В докладе будут вкратце изложены последние достижения в новой области «геномики» болезни Альцгеймера, а также перспективы исследований для разработки диагностики и терапии БА на основе генетических знаний.
1. Роль генетических факторов при болезни Альцгеймера
Еще в 40-х годах была предсказано, что генетические факторы играют значительную роль в патогенезе болезни Альцгеймера как с ранним, так и с поздним началом болезни. Суммируя данные близнецового анализа, характера наследования БА в семьях и недавние результаты анализа генов, вовлеченных в болезнь Альцгеймера, можно сделать следующие выводы:
1) семейные формы с ранним началом БА (до 65, в среднем 35-55 лет) характеризуются аутосомно-доминантным наследованием. Необходимой и достаточной причиной патологии при этом является мутация в единственном гене. Число больных с такими генетическими формами БА, видимо, не превышает 10 % от всех случаев БА.
2) для семейных случаев с поздним началом БА (условно старше 65 лет) наиболее соответствующей характеру наследования является олигогенная (несколько генов) природа наследования с «главной» мутацией в одном или нескольких генах и модификационным эффектом в других генах.
3) «Спорадические» случаи также могут быть обусловлены мутациями или полиморфизмом в генах, патогенная экспрессия которых зависит от многих других генов и эпигенетических факторов.
Обнаружение и клонирование генов БА, в целом, подтверждают данную схему.
2. Гены, мутации в которых ведут к развитию болезни Альцгеймера
К настоящему времени выявлено 4 гена, мутации или вариации, которые бесспорно вызывают или являются факторами риска БА.
Мутации в гене пресенилин 1 ( PS 1, хромосома 14) вызывают наиболее распространенные ранние семейные формы БА и являются, видимо, наиболее «агрессивными» генетическими факторами. Их патологическое проявление характеризуется высокой пенетрантностью и не зависит от других факторов среды или генотипа. К настоящему времени обнаружено более 45 различных миссенс-мутаций, разбросанных по всей длине кодирующей части гена, и одна «сплайсинг» — мутация, ассоциированных с семейной БА. Две мутации гомологичного гена пресенилин 2 ( PS 2, хромосома 1) обнаружены в родословных поволжских немцев и итальянцев.
Анализ многих популяций различного этнического происхождения, в том числе русских (см. в данном сборнике), показал, что полиморфизм в гене аполипопротеина АРОЕ (аллель е4, хромосома 19) является наиболее широко распространенным фактором риска как ранних, так и поздних форм БА. Следует отметить, что в отличие от мутации в гене PS 1 наличие в генотипе индивида е4 варианта не является необходимым или достаточным условием развития БА.
Несколько мутаций в экзоне 16 и 17 у пациентов с ранней семейной БА были ранее описаны для гена предшественника амилоида (APP, хромосома 21).
Суммируя, можно сказать, что мутации в генах пресенилина 1 и 2 ответственны за 30-70 % (по подсчетам разных авторов), а мутации в гене APP менее 5 % случаев семейных форм БА с ранним началом. АРОЕ е4 является одним из факторов риска в 30-50 % всех случаев БА.
В настоящее время продолжается поиск генов, вовлеченных в развитие БА, как методами позиционного клонирования, так и прямым анализом генетической ассоциации полиморфизмов в генах-кандидатах. Для некоторых семейных форм обнаружено «предполагаемое сцепление» БА с геном (еще не идентифицированном) на хромосоме 12. За последние четыре года обнаружено не менее 12 генов, для тех или иных аллельных вариантов которых показаны ассоциации с БА (см., например, в этом сборнике Щербатых и др., Григоренко и др.). К сожалению, известные статистические недостатки метода генетической ассоциации часто не позволяют получать убедительные данные, подтверждаемые в независимых исследованиях.
3. Функции генов болезни Альцгеймера в норме и патологии. Биологическая модель
На первый взгляд, не существует очевидной функциональной или структурной связи между белковыми продуктами тех генов, мутации в которых ассоциированы с БА. Пресенилины — внутриклеточные белки со множественными трансмембранными доменами. APP — белок с одним трансмембранным доменом, локализованным на плазматической мембране. Их экспрессия происходит во всех тканях, в том числе и в нейронах. Аполипопротеин Е — белок со множественными функциями, экспрессируется в мозге, но в глиальных клетках, а не в нейронах.
Исследования клеточных моделей и трансгенных животных показывают, что мутантные формы данных генов изменяют:
1) процессинг APP и аккумуляцию наиболее нейротоксичных форм бета-амилоида;
2) вызывают повышение чувствительности клеток различного типа, в том числе нейронов, к апоптозу;
3) могут влиять на изменение внутриклеточного Са 2+ -гомеостаза и различные системы сигнальной трансдукции и окислительного стресса.
Аполипопротеин Е участвует также в процессах регенерации ЦНС при повреждениях и является фактором риска (е4 форма) при сердечно-сосудистых заболеваниях.
Для выявления непосредственных молекулярных элементов, взаимодействующих с продуктами пресенилинов, APP или АРОЕ, необходимо дальнейшее развитие подходящих клеточных моделей или моделей трансгенных животных. Помимо традиционной модели трансгенной мыши (см. Григоренко и др. в данном сборнике) мы начали создание принципиально новых моделей беспозвоночных (дрозофила и моллюск) для исследования генов БА, и в частности, пресенилинов. В настоящее время клонированы ортологичные гены пресенилинов из Drosophila melanogaster (в сотрудничестве с Университетом г. Торонто) и из улитки Helix L . (неопубликованные данные). Данные модели, как предполагается, будут наиболее подходящими для идентификации элементов сигнальной трансдукции, взаимодействующих с пресенилинами.
С открытием генов пресенилинов остается открытым вопрос: является ли верной «центральная догма бета-амилоида», т. е. бета-амилоид как главная биохимическая причина БА. С одной стороны, показано, что мутации в генах PS 1 и PS 2 и АРОЕ е4 способствуют образованию наиболее «амилоидогенной» формы бета-амилоида. С другой стороны, больные с некоторыми мутациями в генах пресенилинах характеризуются накоплением лишь «диффузных» непатогенных форм амилоидных бляшек. Мутации (Р163Н и H 163 R ) в гене пресенилина 1 способствуют также отложению другого белка — приона (РгР). Наконец, установлено, что подкорковая деменция с множественными инсультами ( CADASIL ) вызывается мутациями в гене Notch 3. В систему Notch -сигнальной трансдукции вовлечен и продукт гена пресенилина. Таким образом, возможно, нарушение системы сигнальной трансдукции в позднем или даже раннем развитии центральной нервной системы может являться фактором ведущим к БА.
Перспективы генной диагностики и терапии
Для проведения генной диагностики БА необходимо: выяснение распространенности мутаций (например, в генах пресенилинов) не только в семейных, но и в спорадических случаях БА и других деменций в разных популяциях; поиск новых генов (например, на хромосоме 12), полиморфизмы в которых являются факторами риска БА или протектирующими факторами при БА.
Уже в настоящее время представляется перспективным тестирование на мутации гена пресенилина 1 у пациентов и родственников в семьях с ранним началом БА. Обнаружение мутации в эволюционно-консервативном сайте пресенилина 1 позволяет, по существу, однозначно предсказать развитие болезни до 60 лет (и наиболее вероятно в возрасте 35-55 лет).
Модели клеточных линий и животных, содержащих трансгенные мутантные формы генов БА, позволят тестировать различные лекарства и молекулы, влияющие на клеточные механизмы
2) апоптоза (програмированной клеточной смерти)
3) сигнальной трансдукции в данных системах.
Это дает надежды на создание лекарственных препаратов, имеющих не симптоматический эффект, а влияющих на первичные молекулярные нарушения при БА.
источник