С Н. Иллариошкин, И А. Иванова- Смоленская. Е.Д. Маркова, ДНК-диагностика и медико-генетическое консультирование в неврологии, 2002
Болезнь Альцгеймера представляет собой одно из наиболее частых нейродегенеративных заболеваний человека и ведущую причину деменции в современном обществе [Rocca W. et al., 1991; Schellenberg G., 1995]. Ее распространенность у лиц старше 65 лет составляет около 3% и неуклонно повышается в более пожилых возрастных группах [Katzman R., 1976]. Заболевание характеризуется прогрессирующим ослабоумливающим процессом, центральное место в развитии которого занимают нарушения памяти, являющиеся наиболее ранним и типичным проявлением болезни. Спустя несколько лет от начала болезни закономерно присоединяются расстройства праксиса, речи, счета, письма, ориентировки и узнавания, у больных могут отмечаться острые психотические эпизоды, эпилептические припадки, разнообразные экстрапирамидные симптомы. В конечном счете развивается глубокая тотальная деменция с распадом личности, тотальная афазия, общее физическое истощение. Средняя продолжительность заболевания составляет около 10 лет. На секции выявляется диффузная атрофия мозга, с большей выраженостью изменений в теменных и затылочных областях. Нри микроскопическом исследовании обнаруживаются два основных гистологических признака болезни Альцгеймера: 1) амилоидные (сенильные) бляшки в паренхиме мозга, главным компонентом которых являются фибриллярные агрегаты гидрофобного пептида (3-амилоида (А[3), состоящего из 39- 43 аминокислот; 2) нейрофибриллярные клубки, представляющие собой спаянные попарно скрученные фи- ламенты измененных нейронов, легко выявляемые при импрегнации серебром [McKee A. et al., 1991]. Характерно также выпадение нейронов, глиоз, наличие единичных телец Леви. Отмеченные изменения наиболее выражены в коре больших полушарий и гиппокампе.
Большинство случаев болезни Альцгеймера имеют мультифакториальную природу и являются спорадическими. В то же время многочисленные популяци опные исследования показали, что 25-40% случаев болезни Альцгеймера могут быть семейными, т.е. в семье пробанда имеется, как минимум, еще один больной с чтим заболеванием [Van Broeckhoven С., 1995]. Важная роль генетических факторов в развитии болезни Альц- I сймера подтверждается высокой конкордантностыо по болезни среди монозиготных близнецов [Breitner J. et al.,
1993]. Анализ большого числа семей с болезнью Альц-
- сймера позволил установить бимодальное распределение значений возраста дебюта симптомов, причем условной границей между ранними и поздними семейны- М и случаями болезни принято считать возраст 58 лет [Van Broeckhoven С., 1995]. В ранних случаях семейной бо- ‘ызни Альцгеймера заболевание обычно наследуется как аутосомно-доминантный признак, связанный с повреждением одного основного гена. Поздние же семейные случаи болезни Альцгеймера являются гетерогенными;
¦ нице всего в этих случаях имеется полигенно обусловленная предрасположенность к болезни Альцгеймера,
- ри которой накопление повторных случаев болезни среди родственников связано с действием комплекса гене- 1 нческих и средовых факторов, общих для членов данной семьи. По некоторым оценкам, наследственные мо- иогенные формы составляют в целом около 5- 10% случаев болезни Альцгеймера [Van Broeckhoven С., 1995; Whitehouse Р.. 1997], и именно им посвящен данный раз- чел монографии. Механизмы генетической предраспо- иоженности и соответствующие методы ДНК-анализа мри ненаследственных формах болезни Альцгеймера подробно разбираются в главе 4, посвященной мульти- фа кториальным заболеваниям нервной системы.
На сегодняшний день идентифицированы 3 гена, м v I ации которых приводят к развитию наследственных (аутосомио-доминантных) форм болезни Альцгеймера с р.шним началом симптомов. Один из них — ген белка- предшественника (3-амилоида, локализованный на хромосоме 21 q21 и обозначаемый аббревиатурой АРР (от англ. Amyloid Precursor Protein) [Kang J. et al., 1987; St George-Hyslop P.H. et al., 1987; Goate A. et al., 1991]. Ген состоит из 19 экзонов, причем аминокислотная последовательность (3-амилоида (А(3) кодируется частью экзонов 16 и 17; данная аминокислотная последовательность расположена в карбоксильной части белка АРР [Kang J. et al., 1987; Yoshikai S. et a]., 1990]. В норме белок АРР подвергается протеолизу под воздействием а-, (3- и у-секретаз; два последних протеолитических пути приводят к высвобождению интактных молекул А(3, что само по себе не сопровождается развитием болезни [Schellenberg G., 1995; Van Broeckhoven С., 1995]. Все восемь известных патогенных толковых мутаций АРР расположены в 16-м и 17-м экзонах гена и ведут к нарушению (3- и у-секретазного процессинга белкового продукта АРР, результатом чего является гиперсекреция пептида А(3 или преимущественная секреция более длинных, склонных к быстрой фибриллярной агрегации форм А(3 [Cai X. et al., 1993; Jarrett J. et al., 1993]. В обоих случаях высвобождаемый пептид А(3 приобретает амилоидогенные свойства-процесс, лежащий в основе формирования сенильных бляшек в паренхиме мозга. Интересно отметить, что мутация Glu693Gln в 17-м экзоне гена АРР приводит к развитию другого заболевания с близким патогенезом — так называемого наследственного церебрального кровоизлияния голландского типа, обусловленного изменением стенок церебральных сосудов вследствие отложения в них амилоидных депозитов [LevyE. et al., 1990]. Более того, оба АРР-ассоциирован- ных заболевания (деменция альцгеймеровского типа и церебральное кровоизлияние) могут наблюдаться у различных родственников в одной и той же родословной — как это описано в семье с мутацией Gly692Ala в 17-м
Зкзоне гена АРР [Hendriks L. et al., 1992]. В целом, мутации в гене АРР представляют собой большую редкость: во всем мире они выявлены лишь в 20 семьях и, по приблизительным оценкам, обусловливают не более 5% всех случаев семейной болезни Альцгеймера с ранним началом симптомов [Van Broeckhoven С., 1995; Blacker D., Tanzi R., 1998].
Два других гена, обусловливающие основную часть случаев ранне-семейной болезни Альцгеймера и расположенные на хромосомах 14q24.3 и lq31-42, были клонированы в 1995 году [Sherrington R. etal., 1995; Levy- Uahad E. et al., 1995; Rogaev E. et al., 1995]. Эти гены являются высокогомологичными и кодируют родственные мембранные белки — пресенилины (соответственно, пресенилин-1 и лресенилин-2). В мозге дресенили- пы экспрессируются преимущественно в нейронах и локализованы в эндоплазматическом ретикулуме тел нейронов и их дендритов [Cook D. et al., 1996; Kovacs D. et al., 1996]. Предполагается, что одна из функций пре- сенилинов может быть связана с регуляцией внутриклеточного транспорта мембранных белков, в том числе белка-предшественника [5-амилоида (АРР) [Hutton М.,
I lardy J., 1997; Sisodia S. et al., 1999]. Мутации в генах прееенилинов сопровождаются гиперпродукцией амилоидогенных форм пептида А[3, формирующих сенильные оляшки [Scheuner D. et al., 1996; Citron M. et al., 1997]. Этот феномен обусловлен, наиболее вероятно, активизацией у-секретазного протеолиза АРР в условиях «задержки» данного белка в эндоплазматическом ретикулуме [Hutton М., Hardy J., 1997]. Другой возможный механизм патогенного эффекта мутантных пресенили- I юв может заключаться в индуцировании апоптоза (про- I раммируемой клеточной гибели) вследствие нарушенной регуляции кальциевого гомеостаза в эндоплазматическом ретикулуме и активизации свободнорадикальных реакций [Mattson М., 1997; Blacker D., Tanzi R., 1998]. В этом случае выявляемое нарушение процессинга АРР в клетках, экспрессирующих мутантные пресенилины, носит вторичный характер по отношению к реализуемому «апоптотическому каскаду». В целом, чуть более половины всех семейных случаев болезни Альцгеймера с ранним началом обусловлены мутациями в генах пресе- нилинов; при этом основная часть случаев связана с пре- сенилином-1 (около 100 описанных семей, gt;50 толковых миссенс-мутаций), тогда как повреждения гена пре- сенилина-2 встречаются весьма редко (лишь 3 описанных мутации) [Blacker D., Tanzi R., 1998; Campion D. et al., 1999]. Следует подчеркнуть, что 70% всех известных мутаций в генах пресенилинов являются уникальными (т.е. каждая из них была выявлена лишь в какой- то одной семье).
Большинство мутаций в генах АРР и пресенилинов характеризуются полной пенетрантностью к концу 6-го десятилетия жизни и неизбежно приводят к манифестации болезни при условии достижения носителем мутации соответствующего возраста. Анализ клиникогенетических корреляций показал отсутствие,каких-либо существенных различий между фенотипами отдельных молекулярных форм болезни Альцгеймера, за исключением возрастных рамок появления первых симптомов болезни. При повреждении гена АРР заболевание манифестирует в возрасте 39-67 лет (чаще всего от 40 до 50 лет), несколько более позднее начало болезни наблюдается у больных с мутациями в гене пресенилина-2 (в среднем от 50 до 65 лет), тогда как в случае мутаций в гене пресенилина-1 заболевание носит наиболее агрессивный и ранний характер (начало болезни от 24 до 56 лет) [Blacker D., Tanzi R., 1998; Campion D. et al., 1999; Lovestone S., 1999]. Некоторые мутации в гене пресенилина-1 могут в единичных случаях вызывать развитие
т ипичного фенотипа болезни Альцгеймера, характеризующегося сочетанием ранней деменции с нижним спастическим парапарезом [Campion D. et al., 1999].
В значительном числе семей с ранней болезнью Альцгеймера мутации в генах АРР и пресенилинов были исключены [Rogaev Е. et al., 1995], что свидетельствует
- Дальнейшей генетической гетерогенности ранней формы заболевания.
Прямая ДНК-диагностика болезни Альцгеймера представляет собой непростую задачу, что связано с генетической гетерогенностью, сравнительно большими
- размерами изучаемых генов и отсутствием в них мажорных мутаций. Опыт такой диагностики в мире имеется
- ишь в сравнительно небольшом числе хорошо оснащен- ных лабораторий, специализирующихся на молекулярно-генетическом анализе данного заболевания. Принимая во внимание, что аутосомно-доминантные формы болезни Альцгеймера характеризуются чаще всего ран-
- км началом заболевания, с практической точки зрения мутационный скрининг целесообразно ограничивать | емьями, в которых хотя бы у части родственников сим- п гомы болезни манифестировали до 60 лет [Campion D. cl al., 1999; Lovestone S., 1999]. Правильному выбору I спа, подлежащего исследованию, может способствовать предварительное установление сцепления болезни с одним из Известных локусов на хромосомах lq, 14q или ’ I q. Если такой анализ сцепления невозможен ввиду небольшого размера обследуемых семей, мутационный ппализ генов проводится в следующей последовательности: пресенилин-1, АРР, пресенилин-2 (в соответствии с частотой встречаемости мутаций в данных генах) |( ampion D. et al., 1999]. Поиск мутаций в пресенили- ппх проводится как на геномной ДНК (SSCP-анализ + прямое секвснирование отдельных экзонов), так и на I ДНК из лимфоцитов крови, которая амплифицируется в виде полного набора перекрывающихся фрагментов и подвергается тотальному секвенированию [Alzheimer’s Disease Collaborative Group, 1995; Campion D. et ah, 1999]. Для ДНК-диагностики мутаций в гене АРР обычно избирательно секвенируются 16-й и 17-й экзоны гена, кодирующие структуру основного патогеннного продукта белка АРР — пептида А(3. При исключении мутаций в исследованных областях всех указанных генов некоторые авторы рекомендую! также секвенировать экзоны 7, 8 и 18 гена АРР, соответствующие функционально значимым участкам АРР-белка [Campion D. et al., 1999]. По данным разных авторов, изучавших различные популяции и выборки больных с ранними аутосомно-до- минантными случаями болезни Альцгеймера, общая частота выявления мутаций в генах АРР и пресенилинов варьирует от 18% до 100% [Plutton М., Hardy J., 1997; Cruts М. et al., 1998; Campion D. et al., 1999]. Поскольку даже в семьях с подтвержденным сцеплением секвени- рование может не выявлять нарушений нуйтеотидного состава кодирующей части гена (это возможно при локализации мутаций в интронах, промоторной области и др.), в таких семьях доступным методом молекулярной диагностики может служить косвенная ДНК-диагностика с использованием маркеров мутантного хромосомного локуса.
источник
Главным представителем нейродегенеративных заболеваний с пораженим коры больших полушарий является болезнь Альцгеймера, которая проявляется деменцией — прогрессирующей утратой когнитивных функций вне зависимости от напряженности внимания. Существует несколько видов деменции: фронтотемпоральная деменция, сосудистая деменция, деменция с тельцами Леви, болезнь Крейтцфельда-Якоба и нейросифилис. При этих заболеваниях также поражаются субкортикальные структуры, но многие симптомы связаны с изменениями в коре больших полушарий. Независимо от этиологии деменция не является нормальным процессом старения, а всегда служит проявлением патологии.
Болезнь Альцгеймера — самая частая причина деменции в пожилом возрасте. Заболевание проявляется постепенными нарушениями интеллекта и расстройствами в эмоциональной сфере и поведении. По мере прогрессирования поражения коры присоединяются дезориентация, мнестические нарушения и афазия. В течение 5-10 лет пациент становится инвалидом, недоступным для контакта и лишенным способности к самостоятельному передвижению.
Симптомы редко возникают до 50 лет, однако частота заболевания с возрастом увеличивается в 2 раза каждые 5 лет: в возрастной группе от 60 до 64 лет частота составляет 1 %, в возрасте от 85 до 89 лет — 40% и выше. Прогрессирующая частота заболевания в связи с возрастом создала множество медицинских, социальных и экономических проблем в странах со стареющим населением. Большинство случаев болезни являются спорадическими, на долю семейной формы приходится лишь 5-10%. Тем не менее изучение именно семейного заболевания пролило свет на патогенез спорадической формы.
Хотя морфологическое исследование ткани мозга по-прежнему необходимо для постановки достоверного диагноза, современная клинико-рентгенологическая диагностика позволяет определить наличие болезни Альцгеймера в 80-90% случаев.
а) Морфология. При аутопсийном исследовании головного мозга видна атрофия коры различной степени выраженности с расширением борозд. Эти изменения особенно заметны в лобной, височной и теменной долях. Из-за выраженной атрофии происходит расширение желудочковой системы (гидроцефалия ex vacuo), поскольку объем мозговой ткани уменьшается. Поражение медиальных отделов височной доли, в т.ч. гиппокампа, энторинальной коры и миндалевидного комплекса, происходит раньше всего, поэтому на поздних стадиях болезни атрофические изменения этих структур выражены особенно сильно.
Главной характеристикой болезни Альцгеймера, позволяющей поставить гистологический диагноз при микроскопическом исследовании, являются сенильные бляшки и сети нейрофибрилл. Наблюдаются прогрессирующая и впоследствии массовая гибель нейронов и реактивный глиоз в местах образования сенильных бляшек и сетей нейрофибрилл.
Сенильные (нейритные) бляшки представляют собой фокальные сферические скопления расширенных и извитых отростков нейронов (дистрофически измененных отростков), расположенных обычно вокруг амилоидного центра, который может быть окружен светлым ободком. Диаметр сенильных бляшек варьирует от 20 до 200 мкм, по периферии располагаются клетки микроглии и реактивные астроциты. Сенильные бляшки обнаруживаются в гиппокампе, миндалевидном комплексе и неокортексе, однако поражение первичной двигательной и соматосенсорной коры выражено в меньшей степени (что также справедливо и для сетей нейрофибрилл).
Амилоидный центр, который можно выявить при окрашивании конго красным, содержит патологический белок. Главный компонент амилоидного центра сенильных бляшек — белок Ар, производное более крупной молекулы — АРР. Аминокислотные последовательности двух основных разновидностей белка Aβ, обозначаемых Aβ40 и Aβ42, имеют одинаковый N-конец и отличающийся на 2 аминокислотных остатка С-конец. Другие белки, присутствующие в сенильных бляшках, малочисленны, к ним относятся компоненты системы комплемента, провоспалительные цитокины, а1-антихимотрипсин и аполипопротеины.
 Болезнь Альцгеймера.
Болезнь Альцгеймера.
Атрофия коры больше выражена в правом полушарии,
где удален лептоменинкс.В ряде случаев происходит отложение белка Аβ со всеми тинкториальными характеристиками амилоида в отсутствие окружающей нейрональной реакции. Эти образования, называемые диффузными сенильными бляшками, обнаруживаются в поверхностных слоях коры больших полушарий, а также в базальных ядрах и коре мозжечка. Диффузное поражение является маркером ранней стадии формирования зрелых сенильных бляшек. Это заключение было сделано при исследовании мозга лиц с трисомией по 21-й хромосоме (синдромом Дауна). Для лиц с синдромом Дауна характерно более раннее начало болезни Альцгеймера.
В некоторых участках мозга (коре мозжечка и стриатуме) диффузные сенильные бляшки сами по себе или наряду с другими изменениями отражают пик развития болезни Альцгеймера. Сенильные бляшки содержат и Aβ40, и Aβ42, а диффузные сенильные бляшки состоят преимущественно из Aβ42.
Сети нейрофибрилл — это пучки филаментов в цитоплазме нейронов, смещающие или окружающие ядро. В пирамидных клетках пучки филаментов внешне часто напоминают рисунок языков пламени; в клетках более округлой формы переплетения волокон вокруг ядра формируют шаровидные сети нейрофибрилл. Эти сети имеют базофильную окраску при окрашивании гематоксилином и эозином и очень хорошо импрегнируются серебром. Сети нейрофибрилл обнаруживаются, как правило, в корковых нейронах, особенно в энторинальной коре, но могут иметь и другую локализацию.
Сети нейрофибрилл встречаются, например, в пирамидных клетках гиппокампа, миндалевидном комплексе, базальных отделах лобных долей и ядрах шва. Сети нейрофибрилл нерастворимы, устойчивы к элиминации и длительно сохраняются после гибели пораженного нейрона в качестве «могильных камней». При ультраструктурном исследовании видно, что сети нейрофибрилл состоят преимущественно из парных спиралевидных филаментов, расположенных вдоль нескольких прямых филаментов, имеющих сходный состав. Основным компонентом парных спиралевидных филаментов являются аномальные гиперфосфорилированные формы тау-белка, который присутствует в микротрубочках аксонов и способствует их соединению.
Другими компонентами являются МАР2 (белок, связанный с системой микротрубочек) и убиквитин. Парные спиралевидные филаменты обнаруживаются также в дистрофически измененных отростках нейронов, которые образуют наружные слои сенильных бляшек, и в аксонах, проходящих через пораженное серое вещество (нити нейропиля). Сети нейрофибрилл выявляются при других заболеваниях, т.е. не специфичны для болезни Альцгеймера.
Болезнь Альцгеймера:
(А) Сенильные бляшки с дистрофическими отростками нейронов вокруг амилоидного центра (стрелки).
(Б) В центре сенильной бляшки и окружающем нейропиле с помощью иммуногистохимического исследования выявляется белок Aβ.Для болезни Альцгеймера характерны и другие патоморфологические изменения. Церебральная амилоидная ангиопатия — неизменный спутник болезни Альцгеймера, но встречается и у лиц, не страдающих болезнью Альцгеймера. При церебральной амилоидной ангиопатии в сосудах откладывается преимущественно Aβ40. Гранулярно-вакуольная дегенерация представляет собой образование мелких (5 мкм в диаметре) светлых вакуолей в цитоплазме нейронов, каждая их которых содержит аргирофильные гранулы.
Вакуоли могут возникать при нормальном процессе старения, но при болезни Альцгеймера такие изменения выявляются преимущественно в гиппокампе и обонятельных луковицах. Тельца Хирано, очень характерные для болезни Альцгеймера, представляют собой удлиненные стекловидные эозинофильные структуры, состоящие из рядов, построенных из паракристаллиновых филаментов, основным компонентом которых является актин. Тельца Хирано обнаруживаются в основном в пирамидных нейронах гиппокампа.
Поскольку изредка сенильные бляшки и сети нейрофибрилл могут быть у лиц, не страдающих деменцией, окончательный диагноз ставят на основании клинических и патоморфологических данных. Прогрессирование болезни идет непрерывно. Патоморфологические изменения (появление сенильных бляшек, сетей нейрофибрилл, гибель нейронов и глиальная реакция) раньше всего появляются в энторинальной коре, затем распространяются через гиппокампальную формацию на мезокортекс и, наконец, достигают неокортекса. Сенильные бляшки оценивают в каждом участке коры (отсутствуют, мало, умеренное количество, много), а сети нейрофибрилл описывают по распространенности в головном мозге.
По этим характеристикам в сочетании с критериями NIA-Reagan устанавливают причастность патоморфологических изменений, типичных для болезни Альцгеймера, к развитию деменции у данного пациента.
Болезнь Альцгеймера.
Сеть нейрофибрилл (вверхуслева) и нейриты вокруг сенильной бляшки (внизу справа),
содержащей тау-белок (иммуногистохимическое исследование).Болезнь Альцгеймера:
(А) Внутри нейронов видны сети нейрофибрилл, которые присутствуют и во внеклеточном пространстве (стрелки).
(Б) При импрегнации серебром видны сети нейрофибрилл в цитоплазме нейронов.б) Молекулярная генетика и патогенез. В основе развития болезни Альцгеймера лежит отложение белков Aβ, которые образуются в результате процессинга АРР. АРР — это поверхностный белок, содержащий один трансмембранный домен. АРР может выполнять функцию рецептора, хотя лиганды этого белка до сих пор не обнаружены. Фрагмент Aβ АРР простирается от внеклеточного пространства до трансмембранного домена. Процессинг АРР начинается с расщепления внеклеточной части домена, затем расщепляется внутримембранная часть домена. Существует два пути, которые различаются по начальной стадии протеолиза. Если первое расщепление происходит в сайте связывания а-секретазы на участке последовательности, соответствующей Aβ, то белок Aβ не образуется (неамилоидогенный путь).
Такой процессинг АРР обычно происходит на поверхности клетки, где различные ферменты с а-секретазной активностью расщепляют поверхностные белки. Поверхностный АРР также может быть подвергнут эндоцитозу, тогда его расщепление происходит с помощью b-секретазы, которая разрывает цепочку аминокислот у N-конца Aβ (амилоидогенный путь). После расщепления АРР в одном из сайтов, у-секретаза осуществляет расщепление внутримембранной части домена. Если первый этап был выполнен а-секретазой, то образуется растворимый фрагмент, если b-секретазой — продуктом расщепления является белок Aβ. Различия в длине белков (Aβ40 vs Aβ42) обусловлены вариабельностью места разрыва аминокислотной цепочки при ее расщеплении у-секретазой. у-Секретазный комплекс, содержащий пресенилин, никастрин, pen-2 и aph-1, также участвует в процессинге сигнальной молекулы Notch, которая наряду с множеством других мембранных белков определяет судьбу клетки.
Белок Aβ имеет очень большую склонность образовывать агрегаты — сначала олигомеры (токсичные для нейронов), затем крупные агрегаты и фибриллы.
Изучение семейных форм болезни Альцгеймера позволило получить доказательства ведущей роли белка Aβ в инициации патологических событий, приводящих к болезни Альцгеймера. Ген, кодирующий АРР, находится на 21-й хромосоме, трисомия по которой ответственна за синдром Дауна. Патоморфологические изменения, характерные для болезни Альцгеймера, обусловливают когнитивные нарушения у лиц с этим синдромом. Характерные для болезни Альцгеймера гистологические изменения появляются в возрасте 10-30 лет, а неврологический дефицит развивается на 20 лет позже. Похожий эффект «умножения генов» наблюдается при локальной дупликации 21-й хромосомы, которая приводит к расширению локуса АРР у некоторых пациентов с семейной формой болезни Альцгеймера.
Точечные мутации гена, кодирующего АРР, тоже могут приводить к появлению семейной формы болезни Альцгеймера. Сайты некоторых мутаций находятся вблизи сайтов связывания b-секретазы и у-секретазы, другие локализуются в пределах последовательности Aβ АРР и усиливают способность белка к агрегации. Два локуса, ответственные за развитие большинства случаев семейной формы болезни Альцгеймера с ранним началом, кодируют два пресенилина: PS1 на 14-й хромосоме и PS2 на 1-й хромосоме. Мутации в этих локусах приводят к появлению патологических функций, например усиленной выработки у-секретазой белка Aβ, особенно Aβ42. Таким образом, генетические данные доказывают ведущую роль белка Aβ в патогенезе болезни Альцгеймера.
Белки Aβ сразу образуют агрегаты, обладающие нейротоксичностью. Есть свидетельства, что мелкие агрегаты вызывают дисфункцию синапсов путем долговременного блокирования передачи синаптических сигналов и изменений свойств других мембран других клеток. Агрегаты очень плохо распадаются, но мономерный Aβ может быть лизирован протеазами. И мелкие, и крупные агрегаты вызывают воспалительную реакцию со стороны микроглии и астроцитов. Не исключено, что эта реакция способствует удалению агрегатов, однако в то же время индуцирует секрецию повреждающих медиаторов. Другими следствиями активации каскада воспалительных реакций являются изменения процесса фосфорилирования и окислительное повреждение нейронов.
Генный локус на 19-й хромосоме кодирует апоЕ, который влияет на риск развития болезни Альцгеймера. Из-за полиморфизма двух аминокислот существуют 3 аллеля белка апоЕ (е2, е3 и е4). Увеличенная порция аллеля е4 повышает риск болезни Альцгеймера и снижает возраст манифестации заболевания. Установлено, что среди всех пациентов с болезнью Альцгеймера носители этого аллеля составляют абсолютное большинство. Аллель е4 вызывает образование белка Aβ и отложение его агрегатов по неизвестному механизму. В целом этот аллель ответственен за 25% случаев спорадической формы болезни Альцгеймера. Вероятно, другие аллели тоже являются факторами риска, но со значительно меньшим влиянием в популяции. В выявлении этих, более слабых локусов может быть полезен внедряемый новый подход к геномному скринингу.
Известно, что сети нейрофибрилл содержат тау-белок, поэтому его роль в развитии болезни Альцгеймера представляет большой интерес. Tay-белок связан с системой микротрубочек аксонов. При образовании сетей нейрофибрилл на фоне болезни Альцгеймера этот белок перемещается в тела и дендриты нейронов, где происходит его гиперфосфорилирование и он утрачивает способность к связыванию с микротрубочками.
Считается, что главным триггером развития болезни Альцгеймера является образование патологической формы белка Aβ, а не влияние тау-белка, поскольку мутации гена, кодирующего белок Aβ, приводят к формированию сетей нейрофибрилл и развитию болезни Альцгеймера, а мутации гена МАРТ, кодирующего тау-белок, вызывают развитие одной из форм фронтотемпоральной деменции, но не накопление белка Aβ. Механизм повреждения нейронов сетями нейрофибрилл мало изучен.
Вопрос о морфологическом субстрате деменции у пациентов с болезнью Альцгеймера остается открытым, однако доказана тесная взаимосвязь широкой распространенности сенильных бляшек и сетей нейрофибрилл с грубыми когнитивными нарушениями, причем количество сетей нейрофибрилл сильнее коррелирует со степенью выраженности деменции, чем количество сенильных бляшек. К биохимическим маркерам, коррелирующим с выраженностью деменции, относятся дефицит холинацетилтрансферазы, иммунореактивность синаптофизина и распространенность отложений амилоида.
в) Клинические признаки. Прогрессирование болезни Альцгеймера — длительный и непрерывный процесс (длительность симптоматического течения часто превышает 10 лет). Начальными симптомами являются мнестические нарушения, по мере развертывания клинической картины заболевания присоединяются речевые расстройства, акалькулия, утрата приобретенного динамического праксиса. Для терминальной стадии болезни Альцгеймера характерны нарушения контроля функции тазовых органов по типу недержания, мутизм и невозможность самостоятельного передвижения. Часто наблюдаются интеркуррентные заболевания (прежде всего пневмония), которые, как правило, приводят к смерти пациента. Одним из важнейших направлений исследований болезни Альцгеймера является выявление специфических биомаркеров. В настоящее время используют позитронно-эмиссионную томографию с амилоидсвязывающим препаратом PiB.
Механизмы процессинга белка-предшественника амилоида.
Существует 2 пути процессинга: неамилоидогенный (расщепление с помощью β-секретазы и y-секретазы) и амилоидогенный (путь, который приводит к образованию агрегатов белка Аβ и амилоидных фибрилл).источник
Болезнь Альцгеймера (БА) — неизлечимая в настоящее время болезнь мозга, получила в 90-х годах XX столетия широкое распространение. По данным ВОЗ, у 1,5% населения земного шара старше 70 лет диагностировано это заболевание и отмечается тенденция к его «омоложению». На долю страдающих болезнью Альцгеймера приходится более половины всех случаев заболевания старческим слабоумием. В США болезнью Альцгеймера поражено 4 млн. человек, по прогнозам в начале XXI столетия эта цифра достигнет 17 млн. человек. Приблизительные прогнозы отмечаются и в других развитых странах. Экономические ежегодные расходы на пациентов пожилого возраста с болезнью Альцгеймера составляют 67,3 млн. дол. (Мах W.,1996). Возрастание доли больных в пожилом возрасте (наивысший уровень заболеваемости наблюдается среди лиц старше 80 лет) объясняется увеличением средней продолжительности жизни, особенно в экономически развитых странах. Болезнь Альцгеймера — прогредиентное заболевание, характеризующее прогрессивным снижением когнитивных функций, утратой способности к правильной оценке окружающего и адекватности поведения. В зависимости от характера сенильно-деструктивных изменений в головном мозге различают следующие варианты деменций: атрофические, сосудистые и смешанные. Болезнь Альцгеймера (деменция альцгеймеровского типа) характеризуется сочетанием диффузного атрофического процесса и локальных очаговых изменений в теменных, височных и лобных долях головного мозга.
Этиология и патогенез болезни Альцгеймера
Этиология заболевания на данный момент не выявлена. Единственным реальным фактором риска считается пожилой возраст. Среди вероятных факторов, способствующих возникновению этого заболевания, отмечаются черепно-мозговые травмы, мини инсульты, возраст матери при рождении пациента старше 30 лет, патология щитовидной железы, аутоимунные заболевания, сахарный диабет, недостаточность ряда витаминов (особенно В12), интоксикация алюминием, вирусная инфекция (трансмиссивные медленные вирусы). Родственники больных, страдающих болезнью Альцгеймера, пациенты с последствиями черепно-мозговых травм, болезнью Дауна и лимфоидозом имеют высокую предрасположенность к заболеванию. У близнецов отмечают высокую конкордантность. Установлен аутосомно-доминантный тип наследования некоторых форм БА, предположительно с 21 парой хромосом, на которой расположен ген белка-предшественника -амилоида. Общность патологии 21 пары хромосом, по-видимому, объясняет развитие характерных для БА изменений головного мозга у пациентов с болезнью Дауна, доживших до 30 лет. Обнаружено также участие в развитии БА 19, 14 и 1 пары хромосом.
Основной патогенетической теорией возникновения БА является нейромедиаторная теория. При БА обнаружены аномалии во многих системах, проводящих импульсы к коре мозга от различных подкорковых образований, эти системы включают холинергическую, норадренергическую и серотонинергическую корковые проекции соответственно от ядра Мейнерта, голубоватого пятна, дорсального ядра шва. Поражение холинергической системы в большей степени обуславливает явления слабоумия, тогда как поражение серотонинергической системы в большей степени способствуют развитию депрессии и нарушений сна. Дефицит в норадренергической системе снижает способность справляться со стрессовыми ситуациями. Дофаминегрическая система при БА относительно интактна.
Поражение преимущественно холинергической системы проявляется снижением содержания в коре холинергических ферментов: холин-ацетилтрансферазы и ацетилхолинестеразы, снижением содержания и синтеза ацетилхолина.
Сниженным оказывается содержание и нейропептида соматостатина. Содержание других собственных нейромедиаторов (глутаминовой, гомованилиновой, гамма-аминомаслянной кислот) находится в пределах нормы.
Морфология при болезни Альцгеймера
Мозг больных, страдающих БА, характеризуется макроскопически резким уменьшением объема и массы мозга (нередко до 900 г), симметричной кортикальной атрофией с сужением извилин коры больших полушарий головного мозга, тотальным расширением борозд при сохранении затылочных зон и области парацентральной извилины, расширением ликворной системы мозга (в частности боковых желудочков).
Окончательный диагноз БА устанавливается после исследования срезов мозга после смерти больного. Подтверждает диагноз наличие большого количества сенильных бляшек в коре и подкорковых ядрах головного мозга в сочетании находящимися в нейронах нейрофибриллярных клубочков. Сенильные бляшки и филаменты состоят из -амилоидного белка, характеризующегося особой -слоистой пространственной структурой. -амилоидный белок, являясь частью нормального липопротеинового комплекса организма, при БА становится нерастворимым и откладывается в образованиях головного мозга в условиях снижения фосфолипид/холестеринового коэффициента и нарушения липид- и липопротеинового синтеза в клетках печени.
Гистологические исследования показывают, что наибольшая гибель нервных клеток отмечается в гиппокампе, базилярной субстанции (ядро Мейнерта). Редукция пирамидных нейронов сопровождается увеличением количества астроцитарных глиальных клеток в неокортикальных зонах и гиппокампе. При БА установлено нарушение структуры систем дендритов пирамидных нейронов, приводящих к нарушению межнейрональной синаптической передачи нервных импульсов и переработки их в коре больших полушарий.
Кроме сенильных бляшек и нейрофибриллярных клубков, характерными для БА гистологическими признаками являются грануловакуолярная дегенерация Смиховича и тельца Гирена. Сенильные бляшки и нейрофибриллярные клубки возникают в результате накопления продуктов нейрофибриллярного перерождения в клетках. Нейрофибриллярные клубки обнаруживаются как в нервных клетках коры больших полушарий, заполняя почти всю цитоплазму и проникая в отростки, так и за пределами клетки — дегенеративный материал, образующийся после гибели нейронов в виде плотных масс грубых нейрофибрилл, остатков ядра, самой клетки. Сенильные бляшки или пластинки состоят из продуктов дегенерации невритов, часто они располагаются вблизи капилляров. В центре пластинки сосредоточен -амилоидный белок — округлой формы ядро, окруженное рядом зон с зернами и палочками, которые состоят из амилоида и могут импрегнироваться серебром и представляют собой продукт дегенерации холинергических нервных окончаний. Обнаруживаются в неокортексе, гиппокампе, а также гипоталамусе, базиллярной субстанции и ядре Мейнерта, в покрышке среднего мозга и верхней части моста.
Грануловакуолярное перерождение — дегенерация Смиховича, представляет собой дегенеративные изменения типа образования в цитоплазме пораженных нервных клеток одной или нескольких вакуолей, в центре которых располагается маленькое базофильной (или аргентофильное) ядро, состоящее из мелкозернистого материала. Встречаются эти образования в клетках гиппокампа, в базиллярной субстанции и миндалевидном ядре.
Тельца Гирена представляют собой палочку, которая состоит из филаментов актина и располагается внутри- и внеклеточно в области гиппокампа. Гистологически при исследовании патоморфологических изменений, обусловленных БА, обращает на себя внимание и мелковакуолярное размягчение поверхностных слоев неокортекса, напоминающее таковое при Крейтцфельдта-Якоба и наличие паравакуолярной амилоидной ангиопатии, которая встречается в 50% случаев БА. Инфильтрация стенок прекапилляров и капилляров коры больших полушарий и мягких оболочек, главным образом в затылочных областях мозга.
Клиника болезни Альцгеймера
По МКБ-10 для диагностики F00 Деменции при болезни Альцгеймера предлагаются следующие диагностические указания:
- Наличие деменции.
- Постепенное начало с медленно нарастающим слабоумием.
- Отсутствие данных клинического или специальных исследований, которые могли бы говорить в пользу того, что психическое состояние обусловлено другими системными или мозговыми заболеваниями, приводящими к деменции.
- Отсутствие внезапного апоплектического начала или неврологических симптомов, связанных с повреждением мозга, таких как гемипарезы, потеря чувствительности, изменение полей зрения, нарушение координации, возникших рано в процессе развития заболевания.
F00.0 Деменция при болезни Альцгеймера с ранним началом (тип2). Деменция при БА с началом до 65 лет с относительно быстро прогрессирующим течением и множественными выраженными расстройствами корковых функций — афазией, аграфией, алексией и апраксией.
F00.1 Деменция при болезни Альцгеймера с поздним началом (тип 1), при которой клинически установленное время начала заболевания после 75 лет и позже. Отмечается медленное прогрессирование с основным симптомом — нарушением памяти.
F00.2 Деменция при болезни Альцгеймера атипичная или смешанного типа.
В отечественной психиатрии выделяют два варианта болезни Альцгеймера: пресенильный и сенильный, в зависимости от возраста начала развития заболевания. По данным Э.Я.Штернберга (1967), начало заболевания до 50 лет наблюдается в 6,5% случаев, от 50 до 60 — в 70,6% случаев, после 60 лет — в 22,9% случаев.
Заболевание в своем развитии проходит через три стадии:
- Начальная стадия (стадия выраженных амнестических расстройств).
- Стадия выраженных очаговых расстройств и выраженного слабоумия (афато-апракто-агностический синдром).
- Терминальная стадия (марантическая или «церебральная смерть»).
В первой стадии болезни на первый план выступают прогрессирующие расстройства предпосылок интеллекта: внимания, темпа функционирования, упражняемости и особенно памяти. Вначале снижается память на события, хронологически близкие к началу заболевания, страдает память времени (больные рассказывают о прошлом, но затрудняются хронологически воспроизвести события), избирательное воспроизведение (припоминание в нужный момент названий, имен, фамилий и др.). У больного развивается амнестическая дезориентировка в месте, времени, чуть позже в собственной личности. Возникает своеобразная растерянность, больные долго сохраняют известную живость и реактивность, известное чувство болезни и измененности. Существует этап истинного критического отношения больных к собственной мнестико-интеллектуальной несостоятельности. Заместительные конфабуляции и сдвиг ситуации в прошлое встречаются редко и исчезают по мере прогрессирования амнестической дезориентировки. Больные становятся безразличны к пробелам в памяти и не пытаются их ничем заполнить. Аутопсихическая дезориентировка может достигать степени неузнавания себя в зеркале.
Очень быстро, быстрее, чем при любом другом виде слабоумия, происходит разрушение всех видов умственной деятельности — способности к суждениям, умозаключениям, абстрагированию и др. Одним из первых проявлений нарушения познавательной деятельности является расплывчатость представлений о характере родственных отношений. В сознании больных родственники объединяются в более обобщенные группы по степени родственной близости, затрудняется точная идентификация отдельных лиц в пределах одной такой группы.
В начальной стадии происходит утрата старых навыков, в первую очередь дифференцированных, тонких и сложных, наиболее поздно приобретенных. Одновременно выявляется и затруднение в восприятии и удержании новых навыков. Утрата больными навыков предшествует развитию грубых апрактических расстройств.
В речевой функции сначала наблюдается тенденция к излишней детализации, затем стремление к объединению предметов, близких по своему предназначению, в более обобщенные группы и определение их наиболее часто встречающимся в обиходе наименованием.
Уже на ранних стадиях заболевания постепенно стирается роль информационной нагрузки общения, происходит сдвиг его в сторону чисто эмоционального реагирования с постепенной утратой синтонности и чувства такта при речевом контакте.
Начальная стадия БА продолжается 3-5 лет. Сравнительно рано происходит постепенное перерастание отдельных компонентов синдрома деменции в очаговые расстройства.
Характерным для второй стадии БА является развитие выраженных очаговых расстройств. Процесс распада совершается послойно: от более «высшего», наиболее сложного и менее закрепленного к более простому. Последовательность возникновения афатических, апрактических и агностических расстройств может быть различной.
Апраксия (конструктивная, идеаторная, моторная) является обязательным признаком БА, начинается она постепенно и малозаметно, параллельно расстройствам памяти и интеллекта. Вначале у больных развивается амнестическая апраксия: нестойкие динамические расстройства праксиса зависят от ситуации, больные лучше справляются при подсказке и руководстве. Расстройства сильнее выявляются при более сложных действиях, касаются плана, последовательности, координации действия, менее страдает возможность манипулирования отдельными предметами. Далее больные забывают не только что надо делать, но и как. Они не справляются с привычными, повседневными профессиональными и домашними делами, становятся несобранными, бестолковыми, беспомощными. В дальнейшем нарушается возможность делать законченные, целенаправленные, адекватные движения, исчезают индивидуальные особенности моторики, выразительные и содружественные движения. Больные не могут одеться, завязать шнурки на обуви.
Часто первым признаком является оральная апраксия, апраксия лицевой мускулатуры: больной не может нахмуриться, высунуть язык.
Постепенно развивается апрактическая обездвиженность: больные не могут ходить, садиться, стоят в неловкой, неестественной позе, топчутся, кружатся. Движения становятся неловкими, неуверенными, незаконченными. Затем наблюдается переход к простым стереотипным, ритмически однообразным движениям, эхопраксия.
Афатические расстройства могут быть ранними или поздними. У больных развивается сначала амнестическая афазия (больные забывают названия предметов, но знают их функцию), затем сенсорная афазия (нарушение понимания речи от сложного до простого, при сохранности повторной речи) и в конце афазия становится тотальной. Часто к сенсорной афазии присоединяется моторная. Происходит нарушение словообразования, мелодики речи. Затруднение и ошибки в произношении слов, расстановке ударений, наблюдаемые еще в конце первой стадии БА, трансформируются в речевые автоматизмы: логоклонии, палилалии, насильственные плач, смех и т.д. Логоклоническое псевдозаикание сменяется насильственным повторением осколков слов на фоне речевого возбуждения, иногда достигающего степени «недержания речи», или аспонтанностью речи.
С прогрессированием болезни развивается агнозия. Вначале появляется пространственная агнозия: больные не ориентируется в новой, а затем и в привычной обстановке, не знают расположения предметов в комнате, окон, дверей, расстояния до них. Они бесцельно бродят, наталкиваются на предметы, бессмысленно трогают и ощупывают все. Развивается и акустическая пространственная агнозия: больные не знают, откуда исходит звук, поворачивают голову не в ту сторону.
На фоне вышеописанных очаговых расстройств развиваются аграфия (нарушение письма), алексия (нарушение чтения), акалькулия (нарушение счета).
Аграфия проявляется в нарушении письма сначала под диктовку, а затем и списывания. Больные пропускают буквы, не заканчивают слов, неразборчиво пишут их, нарушается пространственная структура письма, строки набегают друг на друга, буквы пишутся одна на другой или друг над другом. Далее появляются стереотипии: больные при письме производят стереотипные, затухающие по амплитуде движения, буквы переходят в неразборчивые каракули.
Алексия развивается быстро: от литеральной (больные читают без понимания прочитанного с паралексиями, повторяют чужую речь, не понимая ее) до агностической (больные не узнают букв, не отличают их от других знаков).
Акалькулия выражается в нарушении понятия разрядности чисел, арифметические действия распадаются по мере их сложности: от деления до сложения. Нарушается счет в пределах десятка: вначале абстрактный, затем прикладной. Больные не могут сосчитать пальцы, деньги считают поштучно. Постепенно утрачиваются не только способность к счетным операциям, но и понимание смысла арифметических знаков и значения чисел.
Третья стадия болезни Альцгеймера характеризуется тотальным слабоумием с полным распадом речи, праксиса, узнавания, беспомощностью, нарушением походки, резким повышением мышечного тонуса с развитием контрактур и принятием больным вынужденной эмбриональной позы. Наблюдаются разнообразные насильственные двигательные, оральные и хватательные, автоматизмы, насильственные гримасы плача и смеха. Развиваются эндокринные нарушения (маскулинизация у женщин), похудение до кахексии, иногда на фоне булимии и пристрастия к несъедобным вещам, крайнее одряхление. Часто присоединяются различные соматические заболевания, являющиеся причиной гибели этих больных.
БА может сопровождаться различными неврологическими расстройствами: паркинсоноподобные (акинетически-гипертоническими) проявлениями, хорее подобными и миоклоническими гиперкинезами.
Эпилептические припадки встречаются в 30% случаев заболевания, возникают на разных стадиях заболевания, чаще через 5 и более лет от начала БА. При раннем начале заболевания они возникают в 9 раз чаще (Lauter H.,1970). Чаще припадки бывают единичными, возникают как типично генерализованные, так и абортивные состояния типа обмороков с подергиваниями. Редко встречаются кратковременные сумеречные состояния.
Помимо описанных когнитивных нарушений при БА наблюдается группа некогнитивных нарушений, которую в свою очередь можно подразделить на две подгруппы (Reisberg B.,1983): психопатологические симптомы и собственно нарушения поведения. К первой подгруппе относятся бредовые расстройства, галлюцинации, нарушение ритма сна и бодрствования, тревога, страх, нарушения аффекта.
Бредовые расстройства в виде нестойких, отрывочных бредовых идей возрастного содержания возникают на начальной стадии. Они не трансформируются в другие синдромы, а постепенно распадаются. Специфичным для БА является бред обкрадывания.
Острые кратковременные психотические эпизоды в виде галлюцинаторной спутанности, делириозных расстройств свидетельствуют об ускорении темпа атрофическим процессом зрительной области.
Тревога возникает при необходимости претерпеть какую-либо встречу или событие. Довольно частым явлением бывают страхи: страх остаться одному, страх толпы, темноты, принятия ванны и т.п. Эти симптомы возникают на фоне атрофического процесса. На поздних стадиях иногда наблюдаются отдельные отрывочные галлюцинации и скоропреходящие рудиментарные галлюцинозы без нарушения сознания, чаще зрительные. Это является признаком коркового поражения на разных стадиях заболевания, и не обнаруживают какой-либо связи с прогрессированием заболевания.
Аффективные расстройства довольно часто встречаются при БА. Хотелось бы отметить, что депрессивные расстройства часто могут усугублять, а иногда и имитировать снижение уровня личности, даже амнестические и афатические нарушения. Поэтому необходимо использовать весь комплекс диагностических и дифференциально-диагностических критериев при диагностике слабоумия. Это представляется более важным, чем использование тестовых оценок функции памяти, мышления и речи для заключения о наличии деменции (Шахматов Н.Ф.,1998).
Эта подгруппа некогнитивных психопатологических расстройств довольно хорошо поддается медикаментозной коррекции.
Вторая группа некогнитивных расстройств представлена нарушениями поведения. В известной мере нарушения поведения связаны с прогрессированием заболевания. К ним относят: агрессивность, ажитацию, беспокойство, расторможенность, блуждания, бесцельную активность (суетливость, бессмысленное хождение «туда-сюда»), нарушения активности (больные накапливают разные предметы или прячут свои вещи в неподходящих местах) и др. Нарушения поведения практически не поддаются медикаментозной коррекции, что делает особо важной правильную организацию ухода за такими больными.
По темпу прогрессирования и степени выраженности синдромов выделяют два варианта течения заболевания. Первый наблюдается в пресенильном возрасте и характеризуется быстрым развитием деменции и присоединением очаговой симптоматики на самых ранних этапах болезни. При втором варианте атрофический процесс начинается в более позднем возрасте, характеризуется медленным прогрессированием и значительно более поздним присоединением очаговой симптоматики.
Методы диагностики болезни Альцгеймера
Для диагностики БА используются следующие методы:
- клинический психопатологический метод;
- катамнестический метод;
- нейропсихологический метод;
- инструментальные методы, в том числе а) ЭЭГ, б) КТ, в) МЯР, г) ПЭТ, д) SPECT.
В качестве дополнительных методов, используемых для дифференциальной диагностики БА, применяются реоэнцефалография, ультразвуковая доплерография, биохимический (уровень ацетилхолина, норадреналина, СТГ, тироксина, эстрогенов, и др.), иммунологический и генетический методы.
Клинический метод предусматривает опрос больного и его ближайших родственников, осмотр пациента, формирование предварительного диагноза и плана дальнейшего обследования, терапевтическую тактику.
Катамнестический метод — метод лонгитудинального наблюдения за больным — предоставляет возможность уточнения диагноза и предположения прогноза заболевания на основе динамики проявлений болезни. Появление новой симптоматики, характерной для БА, а также отсутствие резких противоречий между отдельными сторонами клинической картины помогают верифицировать диагноз.
Нейропсихологическое исследование особенно полезно, т.к. дает возможность относительно ранней диагностики, а также дифференциальной диагностики и оценки степени тяжести заболевания. Используются методики исследования памяти, внимания, интеллекта (на традициях синдромального анализа школы А.Р.Лурия), самооценки (методика Дембо-Рубинштейна), депрессии (методики Гамильтона, Бека), а также оценочные шкалы: клиническая гериатрическая шкала Сандоз (Sandoz Clinical Assessment Geriatric (SCAG) scale) и методика мини-исследования психического состояния (Mini Mental State Examination, MMSE), шкала глобального ухудшения психических функций (GDS).
В последнее время все большее предпочтение в диагностике БА отдается инструментальным методам.
Электроэнцефалография (ЭЭГ) не выявляет патологических изменений на ранних этапах болезни, в дальнейшем отмечается симметричное диффузное замедление мозговой активности.
Компьютерная томография мозга (КТ) и магнитно-ядерный резонанс (МЯР) также не дают возможности для ранней диагностики БА. На поздних этапах страдания выявляются атрофия извилин, расширение борозд и компенсаторное расширение желудочков мозга. С помощью МЯР можно обнаружить также уменьшение слоя серого вещества. Сочетанное применение этих методов особенно информативно в случаях дифференциальной диагностики БА и других причин деменции (мультиинфарктная деменция и объемные процессы ).
Сканирование методом позитронно-эмиссионной томографии (ПЭТ) позволяет выявить уменьшение поглощения корою мозга глюкозы. При БА оно больше выражено в теменных участках, меньше в височных и лобных.
SPECT (однофотонная эмиссионная компьютерная томография) выявляет одно- или двух стороннее снижение кровенаполнения в теменных и височных областях мозга. Этот метод позволяет дифференцировать БА с болезнью Пика, при которой кровенаполнение снижено в лобных участках мозга.
Наиболее достоверным диагноз становится после посмертной аутопсии мозга с морфологическим исследованием тканей неокортекса. Наличие характерных морфологических признаков (сенильные бляшки, нейрофибриллярные клубки, грануловакуолярная дегенерация Смиховича и тельца Гирена) делает диагноз болезни Альцгеймера окончательным.
Для установления диагноза болезни Альцгеймера необходимо провести дифференциальную диагностику клинической картины заболевания пациента с деменциями, вызываемыми другими причинами: сосудистой деменцией, болезнью Пика, хореей Гентингтона, болезнями Крейтцфельдта-Якоба и Паркинсона, деменцией при объемном процессе головного мозга. Снижение, а иногда и имитацию усугубления интеллектуально-мнестических функций и уровня личности могут вызывать депрессивные расстройства, возникающие у пожилых лиц. Эти состояния («псевдодеменция») также входят в круг состояний для дифференциальной диагностики БА.
По МКБ-10 F01 Сосудистая деменция имеет отличительные особенности. В типичных случаях отмечаются преходящие ишемические эпизоды с кратковременной потерей сознания, нестойкими парезами, потерей зрения. Деменция также может наступать после серии острых цереброваскулярных эпизодов, или после одной большой геморрагии. Когнитивные нарушения обычно неравномерные (парциальные) — потеря памяти, интеллектуальное снижение, очаговые неврологические знаки. Различают: F01.0 Сосудистую деменцию с острым началом, развивающаяся после серии инсультов, цереброваскулярного тромбоза, эмболии или геморрагий. F01.1 Мультиинфарктную деменцию, характеризующуюся постепенным началом с «накоплением» очагов — инфарктов в церебральной паренхиме. F01.2 Субкортикальную сосудистую деменцию, возникающую вследствие гипертензии, очагов ишемической дисфункции в глубоких структурах белого вещества. Клиника субкортикальной сосудистой деменции напоминает таковую при болезни Альцгеймера, различия очевидны по результатам компьютерной аксиальной томографии. F01.3 Смешанная корковая и подкорковая сосудистая деменция. Сосудистые заболевания головного мозга могут приводить к развитию слабоумия, сходного по клиническим проявлениям с БА. Однако сущeствуeт ряд признаков, указывающих на сосудистую природу заболевания: наличие в анамнезе указаний на артериальную гипертензию, эпизоды острого нарушения мозгового кровообращения с развитием после них афатических, агностических и апрактических расстройств, наличие двигательных нарушений в виде парезов и параличей, изменений чувствительности, очаговой неврологической симптоматики, псевдобульбарного синдрома, гиперкинезов, дрожательного паралича. Мультиинфарктная деменция может возникать достаточно рано, чаще у относительно молодых мужчин со склонностью к гипертензии, частыми психоэмоциональными стрессами в анамнезе. Атеросклеротическое слабоумие развивается по мере прогрессирования атеросклеротического процесса в сосудах головного мозга. Течение процесса неровное, ступенеобразное, что объясняется сохраняющейся возможностью к компенсации. Характерным для сосудистой деменции является отсутствие присущей БА последовательности развития расстройств высших корковых функций. Вся симптоматика сосудистой деменции носит на себе налет болезненности и слабости, как на продуктивной, так и на дефицитарной симптоматике. Характерными для начального этапа сосудистой деменции являются жалобы на неспособность к сосредоточению, несобранность, снижение трудоспособности, гиперестезию, а в дальнейшем на неспособность к умственному напряжению вообще. Заостряются личностные черты при относительной сохранности ядра личности. Возникают нарушения мышления в виде его замедления, тенденции к обстоятельности, склонность к рассуждательству. Аффективные реакции становятся неустойчивыми, возникает недержание аффекта, слабодушие, явления насильственного плача. Возникающая продуктивная симптоматика, как правило, монотонна, маловыразительна. Частичное осознание собственной несостоятельности, неполная потеря ориентировки в окружающем, элементы адекватности в эмоциональных реакциях позволяют говорить о парциальном характере слабоумия при сосудистой деменции. Относительным дифференциально-диагностическим критерием сосудистой деменции является положительный ответ на проведение курса сосудистой терапии (сосудорасширяющие, ноотропные, гипотензивные, холестеринснижающие средства, антикоагулянты и др.).
F02.0 Деменция при болезни Пика обнаруживается рано и также, как и при БА, неуклонно прогрессирует. От БА отличается более ранним началом (в возрасте 50-60 лет). Социальные и поведенческие нарушения при болезни Пика часто предшествуют нарушениям памяти. В большей степени выражена неврологическая симптоматика. Патоморфологические отличия от БА – преобладание глиоза, массивные клеточные потери в лобных и теменных долях, наличие патогномоничных телец Пика и отсутствие характерных для БА нейрофибриллярных изменений и сенильных бляшек.
F02.1 Деменция при болезни Гентингтона. Хорея Гентингтона — наследственное заболевание, возникающее в возрасте 30-50 лет. Начальными проявлениями заболевания являются хореоатетоидные двигательные расстройства, которые часто ошибочно принимаются за спазмы или тики, депрессия, тревога, бред и изменения личности. На более поздних стадиях возникает деменция, как правило, с психотической симптоматикой. Она относительно мало прогредиентна. Частично сохраняется трудоспособность наряду с несостоятельностью в непривычной деятельности. Расстройства памяти начинаются с затруднения запоминания и репродукции, не достигают степени, наблюдающейся при БА. Ориентировка во времени и собственной личности нарушается редко. Происходит интеллектуальное снижение, обеднение психической деятельности. Течение медленно прогрессирующее, смерть наступает через 15-25 лет.
F02.1 Деменция при болезни Крейцфельдта-Якоба, которая представляет собой быстро прогрессирующее дементивное заболевание, вызываемое вирусом медленной инфекции. Чаще возникает в возрасте 40-60 лет. Стремительность развития деменции (в течение 1-2 лет), наличие массивной неврологической симптоматики (пирамидные и экстрапирамидные парезы и параличи, миоклонии, ухудшение слуха, нарушения походки, эпилептиформные припадки), характерная трехфазная ЭЭГ являются отличительными признаками этого заболевания. Возможна продуктивная симптоматика (эпизодические помрачения сознания со слуховыми галлюцинациями, конфабуляции).
F02.3 Деменция при болезни Паркинсона. При этом заболевании интеллектуально-мнестическое снижение на фоне легкой эйфории возникает на поздних стадиях заболевания. Болезнь Паркинсона развивается в позднем возрасте и проявляется в основном экстрапирамидными расстройствами в виде брадикинезии, тремора, симптома «скатывания пилюль», маскообразной мимики, мышечной ригидности по типу зубчатого колеса, шаркающей походки. К неврологическим расстройствам присоединяются характерные изменения личности: повышенная раздражительность, эгоцентризм, назойливость, подозрительность. Мнестико-интеллектуальные расстройства часто сочетаются с депрессивными нарушениями. Успешная коррекция состояния пациентов с помощью средств, влияющих на уровень дофамина в головном мозге (леводопа, амантадин, юмекс и др.), также является дифференциально-диагностическим признаком этого заболевания.
F02.4 Деменция при заболеваниях, обусловленных вирусом иммунодефицита человека (ВИЧ), обычно характеризуется жалобами на забывчивость, медлительность, трудности в концентрации внимания, решении задач и чтении. Нередки апатия, снижение спонтанной активности и социальная отгороженность. В некоторых случаях отмечаются атипичные аффективные расстройства, судорожные пароксизмы и психотические состояния (делирий, острые параноидные психозы, галлюцинозы). Неврологическое обследование выявляет тремор, нарушение быстрых повторных движений, координации, атаксию, повышение рефлексов, лобное растормаживание и нарушение глазодвигательных функций. Деменция при ВИЧ может прогрессировать в течение недель и месяцев до уровня маразма и смерти.
Основным отличием деменции, возникающей при опухолях головного мозга, является сочетание общемозговой симптоматики и очаговых неврологических расстройств, соответствующих локализации объемного процесса. Темпы развития деменции и ее клинические проявления зависят от локализации опухоли. Большую роль в дифференциальной диагностике этих состояний играют дополнительные методы исследования (Эхо-ЭГ, КТ и МЯР).
Часто приходится проводить дифференциальный диагноз между деменцией и депрессией. При депрессии больной тоже может быть апатичен, заторможен, у него нарушена способность к сосредоточению, замедлено мышление, снижена мотивация, снижены интеллектуально-мнестические способности. Однако после выхода из болезненного состояния все эти функции восстанавливаются. Важным дифференциально-диагностическим признаком деменции при БА и депрессии является обратимость деменции под влиянием курса лечения антидепрессантами.
б) лечение сопутствующих депрессивных расстройств
в) лечение психотических проявлений
г) медикаментозная коррекция поведенческих нарушений
д) психологические меры коррекции изменений в поведении больных
е) консультирование и поддержка семей, в которых живут больные
3. Консультирование ближайших родственников больных БА с целью ранней диагностики возможного заболевания.
Современные подходы к медикаментозной коррекции клинических проявлений БА основываются на этиопатогенетических гипотезах развития данного заболевания. Используются лекарственные средства, оказывающие влияние на функцию головного мозга: гамма-аминомасляная кислота (ГАМК) и ее производные, алкалоиды спорыньи, нейропептиды, предшественники нейромедиаторов, препараты, предупреждающие церебральную аноксию, снижающие вязкость крови, мембраностабилизаторы, а также средства, активирующие процессы обмена в головном мозге.
Существует несколько направлений в терапевтической стратегии БА. Во-первых, усиление высвобождения нейротрансмиттеров, в частности ацетилхолина, как наиболее истощаемого трансмиттера, а также серотонина, норадреналина и некоторых нейропептидов (соматостатин, вазопрессин, тиреотропин-высвобождающий фактор). Повышение уровня нейротрансмиттеров имеет целью общее симптоматическое повышение их уровня без намерения изменить прогрессирование болезни. Во-вторых, замедление и прекращение дегенерации нервных клеток. Применение противовоспалительных средств (ацетилсалициловая кислота), соединений алюминия, блокаторов кальциевых каналов и ингибиторов моноаминооксидазы типа В (МАО-В) является примером этого подхода. И, наконец, в-третьих, предпринимаются активные попытки спасти погибающие нейроны и стимулировать восстановление синаптических связей путем увеличения факторов роста нервов.
Для увеличения содержания ацетилхолина в пресинаптическом окончании использовались предшественники медиатора — холин и лецетин. Большой интерес вызывает применение ингибиторов ацетилхолинэстеразы (нейромедин, физостигмин, такрин, амиридин). Два последних препарата являются одновременно блокаторами калиевых каналов, что обеспечивает выход большего количества ацетилхолина в синаптическую щель. Ингибиторы ацетилхолинэстеразы обладают способностью снижать отложения бета-амилоида в ткани мозга. Нейромедин, Такрин и Амиридин улучшают когнитивные функции, облегчают симптомы деменции, при длительном применении снижают темп прогредиентности заболевания.
Холинергическая передача в головном мозге осуществляется преимущественно мускариновыми рецепторами. Никотиновые холинергические рецепторы также страдают при БА. Обнаружено, что лица, когда-либо курившие сигареты, имеют более низкий риск заболеть БА. Никотин защищает холинергические и дофаминергические рецепторы. Применение никотина вызывает улучшение внимания, обучения, сокращение времени реакций и решения задач, улучшает скорость переработки информации. У больных БА отмечалось значительное улучшение внимания, восприятия, скорости реакций, поведения, изменения когнитивной функции не наблюдалось.
С точки зрения коррекции других медиаторных систем, страдающих при БА, эффективным является препарат юмекс (селегелин, депренил). Он является избирательным ингибитором МАО-В, активность которой повышена при БА. Препарат стимулирует также выделение катехоламинов, в том числе дофамина, повышает активность ферментов, инактивирующих свободные радикалы, (активность ферментов при БА снижается). Применение юмекса способствует редукции депрессивной симптоматики, уменьшению тревоги и напряженности у больных БА, увеличивает их активность, улучшает когнитивные функции, социальные взаимодействия, способность к решению сложных задач. Эффективно применение препарата при нарушении когнитивно-познавательной сферы и поведенческих реакций у больных на ранних и средних стадиях БА, т.к. он замедляет прогрессирование заболевания. Юмекс в сочетании с ингибиторами ацетилхолинэстеразы составляет базовую терапию БА и деменций альцгеймеровского типа.
Триптофан и тирозин являются предшественниками дофамина, норэпинефрина и серотонина. Концентрация этих веществ снижена в тканях мозга уже на ранних стадиях БА. Низкий уровень триптофана и тироксина и высокий уровень цистеина в плазме крови отражают наличие изменений белкового метаболизма при старении и БА. Эти данные говорят о целесообразности включения в коррекционную терапию растворимых предшествеников триптофана и тирозина.
При БА повышается содержание свободного кальция в нервных клетках. Нейротоксичность бета-амилоида проявляется в дестабилизации кальциевого гомеостаза. Для предотвращения избыточного накопления внутриклеточного кальция используются блокаторы кальциевых каналов. Представителями этой группы препаратов являются нимодипин и ницерголин (сермион). Эти препараты улучшают когнитивные функции, способность к концентрации внимания и работоспособность больных, длительная терапия способствует замедлению патологического процесса.
Нейротоксичность алюминия, его способность вызывать агрегацию бета-амилоида, снижение когнитивных функций и повышение частоты БА при применении питьевой воды с содержанием алюминия способствовали поиску лекарственного препарата, связывающего ион алюминия. Таким средством является десферриоксамин. Отмечена способность препарата уменьшать прогрессивность заболевания. Десферриоксамин связывает и железо, благодаря чему уменьшает повреждения, вызванные свободными радикалами, обладает противовоспалительными свойствами.
Нейротрофические факторы (НТФ) — это секреторные белки, действующие непосредственно на нейроны, регулирующие их долговременное переживание, вызывающие дифференциацию, защищающие нейроны от последствий различных повреждений и поддерживающие их регенерацию. Используются NGF — фактор роста нервов и эстрогены. Эстрогены усиливают образование нейрональных рецепторов для фактора роста нервов. В этих же целях используют церебролизин — пептидергический ноотропный препарат. Церебролизин проявляет свойства улучшать метаболическую регуляцию, нейропротекцию, проявлять нейротрофическую активность аналогично действию естественных факторов роста нервов, осуществляет функциональную нейромодуляцию. У больных, страдающих БА, использование этого препарата вызывает улучшение общего состояния и когнитивного функционирования, способствует уменьшению расстройств двигательных, интеллектуальных и эмоциональных функций даже после прекращения введения препарата (эффект последействия).
Применение препаратов — церебральных вазодилататоров и нейрометаболитов, таких как кавинтон, циннаризин, инстенон, эуфиллин, пирацетам, аминалон, энцефабол и др., способствует улучшению кровоснабжения головного мозга, улучшению утилизации глюкозы клетками мозга, блокированию биологически активных веществ спазматического действия. Это улучшает способность больных БА к выполнению психометрических тестов, касающихся параметров познавательной деятельности, улучшает память, способность концентрировать внимание, работоспособность.
Для коррекции депрессивных, тревожных, психотических расстройств, бессонницы, нарушений поведения, возникающих у больных БА, используются антидепрессанты, нейролептики, транквилизаторы, корректоры сна и поведения.
Целый ряд нарушений поведения при БА не поддается психофармакотерапии. В связи с этим большое значение придается правильной организации факторов окружающей среды, способствующей уменьшению поведенческих нарушений. К ним можно отнести: наличие достаточно просторного и безопасного помещения, где больные имели бы возможность двигаться, так как стеснение часто приводит к нарушениям поведения, поддержание привычной, неизменной, не угрожающей и предсказуемой обстановки, избегание больших «пустых» промежутков времени в распорядке дня, проведение разъяснительной работы с лицами, осуществляющими непосредственный уход за больным.
Основные терапевтические мероприятия у пациентов с сосудистой деменцией должны быть направлены на основной патологический процесс, улучшение церебральной гемодинамики и повышение функциональных возможностей головного мозга. Основными причинами сосудистых (дисциркуляторных) энцефалопатий и деменций являются гипертоническая болезнь и атеросклероз. В лечении гипертонической болезни за последние годы достигнуты значительные успехи. Адекватное сочетание антигипертензивных препаратов с диуретиками, транквилизаторами, адреноблокаторами и антидепрессантами в большинстве случаев приводит к удовлетворительным результатам. Перспектива лечения атеросклероза менее утешительна.
Воздействие на основной патологический процесс
Из препаратов, используемых для лечения атеросклероза, следует отметить гиполиподемические средства: полиспонин, трибуспонин, клофибрат, цетамифен, арахиден, зокор, мевакор.
Вторым компонентом патогенеза атеросклероза является сосудисто-тромбоцитарный механизм. При атеросклеротической энцефалопатии наиболее часто используются ацетилсалициловая кислота (каталгикс) и дипиридамол (курантил). В последнее время более эффективным препаратом признан тиклид — истинный антиагрегант прямого механизма действия. Тиклид мощный ингибитор пластиночной агрегации, индуцированной АТФ. Максимальный эффект тиклида, назначаемого в суточной дозе 500мг, наблюдается после 8-11 дней приема. Возможно длительное применение. Для профилактики церебральных ишемий тиклид назначается сроком от1 до 3-х месяцев (Кузнецова С.М., 1997).
Перекисная теория является основанием для использования антиоксидантов в лечении атеросклероза. Препараты, обладающие антиоксидантной активностью, делятся на прямые антиоксиданты (аскорбиновая кислота, флавонаноиды, токоферол), и непрямые антиоксиданты — вещества, повышающие антиоксидантный потенциал тканей (метилметионин, липоевая и глутаминовая кислоты, никотинат ксантинола).
Улучшение мозговой динамики — второе направление в лечение сосудистой энцефалопатии и деменции. Клиническая практика убедительно показано преимущество средств, сочетающих сосудорасширяющее и метаболическое действие. К таким препаратам относятся винкамин, винкатон (винкапан) и винпоцетин (кавинтон), циннаризин, трентал, стугерон. Кавинтон является оптимизатором мозгового кровообращения, а у винкамина и винкатона больше выражен антигипертензивный эффект. Из этих препаратов чаще всего применяется кавинтон. Вазомоторный эффект препарата сложный и осуществляется по нескольким механизмам: увеличением синтеза катехоламинов и АТФ, активацией энергетических процессов в головном мозге. Назначается препарат в дозе 10 мг 3 раза в день. Курс лечения 3-4 месяца.
Пентоксифиллин (Трентал) повышает уровень АМФ, регулирует функцию К-насоса клеточных мембран, потенцирует влияние -адренергических агонистов. Фармакологическая эффективность препарата обусловлена влиянием на венозное и артериальное кровообращение. Трентал назначается в дозе 100 мг 3 раза в день в течение 2 месяцев.
Исследования 90-х годов показали высокую эффективность ницерголина (Сермиона) в лечении сосудистой деменции (Белоусов Ю.Б. и др., 1997). Ницерголин оказывает -адреноблокирующее действие, а также активирует метаболизм, снижает сопротивление сосудов мозга, усиливает артериальный кровоток, увеличивает потребление кислорода и глюкозы тканями мозга. Для лечения деменции препарат назначается по 5-10 мг 3 раза в сутки, лечение длительное, не менее 3-х месяцев.
Широкий спектр вазоактивного действия антагонистов кальция определяет перспективы применения этой группы препаратов в лечении сосудистой деменции.
Нифедипин (адалат) обладает спазмолитическим и нейрометаболическим действием. Положительное влияние препарата на клиническую симптоматику и состояние церебральной гемодинамики обуславливает его применение. Нифедипин увеличивает кровоток в ишемизированных участках мозга и не вызывает симптомов обкрадывания в здоровых. Нифедипин назначают по 20-40 мг в сутки. При выраженной гипотензии препарат назначать не следует, особенно при систолическом артериальном давлении ниже 90 мм рт.ст. Максимальная суточная доза 60 мг, курс лечения 1-2 месяца.
Нимодипин (нимотоп) показан при сосудистых деменциях с жалобами на нарушение памяти, мотивации, концентрации внимания, лабильности настроения. Рекомендуемая суточная доза 3 раза по 1 таблетке (30мг нимодипина). Длительность лечения обычно составляет несколько месяцев.
Для лечения больных с деменцией применяются вазоактивные препараты растительного происхождения: из алколоидов спрыньи (редергин), экстракт Гинкго билоба (танакан). Перспективно использование комплексных гомеопатических лекарственных средств (Церебрум композитум).
Основным направлением в лечение деменции является применение препаратов, улучшающих метаболизм мозга. Все вышеперечисленные препараты обладают свойством улучшать метаболизм, однако имеется специфическая группа препаратов — ноотропов: пирацетам, пиридитол, пикамилон, церебролизин, ноофен, олантропин, актовегин.
Пирацетам применяется внутрь по 400-800 мг 3 раза в день. Продолжительность лечения 1-3 месяца.
Ноофен назначают по 250-500 мг 3 раза в день. Курс лечения 4-6 недель.
Олатропил – комбинированный препарат, в состав которого входят аминалон и пирацетам. Учитывая сходные и различные эффекты пирацетама и аминалона, их совместная комбинация дает возможность нивелировать побочные эффекты друг друга, потенцировать положительное действие и позволяет, повышая эффективность лечения, снизить терапевтическую дозу каждого из компонентов. Олатропил применяется по 1 капсуле 3-4 раза в день, курс лечения 4-8 недель.
Пиридитол (энцефабол) улучшает патологически сниженные обменные процессы в ишемизированных зонах мозга, что связано с повышением усвоения и метаболизма глюкозы, обмена нуклеиновых кислот, а также повышением высвобождения ацетилхолина и активацией холинергических процессов. Препарат назначают в дозе 100 мг/сут. в течение 2-х месяцев.
Пикамилон – метаболический цребровазорегулятор и ноотроп назначается внутрь по 0,02-0,05 г 3 раза в день. Курс лечения 1-2 месяца, при необходимости через 5-6 месяцев курс лечения можно повторить.
Церебролизин — очищенный от белка гидрализат мозговой ткани, содержит 18 аминокислот и полипептиды. Церебролизин существенно замедляет или даже приостанавливает прогрессирование нейродегенерации, регулирует внутриклеточный синтез белков, положительно воздействует на пластичность нейрональных мембран и облегчает транссинаптическую передачу. При лечении церебролизином достоверно отмечается улучшение абстрактного и практического мышления, памяти, внимания. В последнее время появились сообщения о применении существенно больших, чем общепринятые, доз — от 10 до 30 мл внутривенно. По данным разных авторов препарат хорошо переносится и не оказывает побочного действия.
Актовегин — депротеинизированный гемодериват из телячьей крови. Улучшает аэробный энергообмен нервной клетки, повышает метаболизм глюкозы, стимулирует окислительные процессы, улучшает микроциркуляцию и венозное кровообращение. В комплексном лечении сосудистых деменций назначается по 2,0 мл в/м на курс 15-25 инъекций.
Помимо медикаментозного лечения при сосудистой деменции проводят психотерапию, лечебную физкультуру, трудотерапию и другие реабилитационные мероприятия.
Глава 13. ГИНЕКОЛОГИЧЕСКАЯ ПСИХИАТРИЯ
источник
 Болезнь Альцгеймера.
Болезнь Альцгеймера. 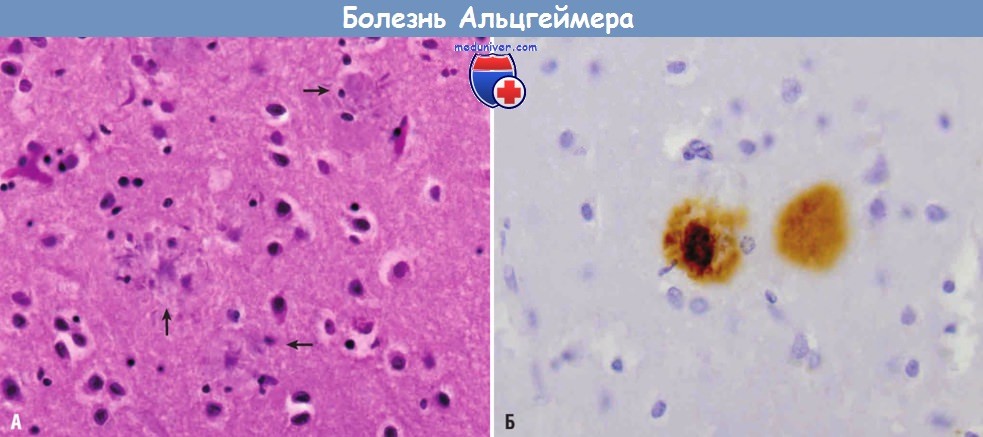 Болезнь Альцгеймера:
Болезнь Альцгеймера: 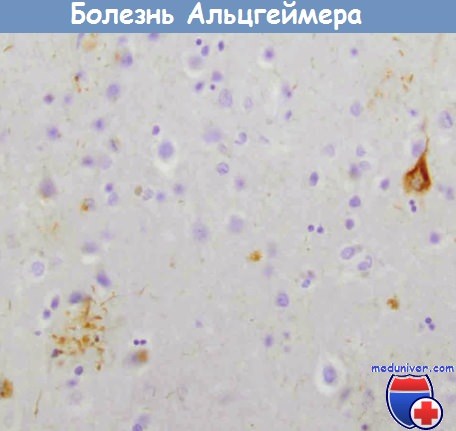 Болезнь Альцгеймера.
Болезнь Альцгеймера. 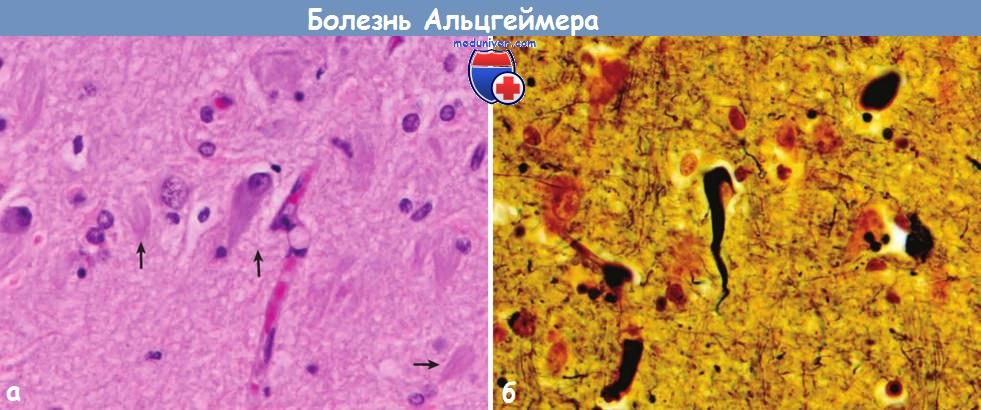 Болезнь Альцгеймера:
Болезнь Альцгеймера: